The Transportome of the Human Mucosal Pathogen Ureaplasma urealyticum
Sudhakar, Padhmanand
St.Peter's Engineering College Chennai, , India
Subramani, Prasanth
St.Peter's Engineering College Chennai, , India
Abstract
has been one of the most prominent mucosal pathogens having many tropisms in humans. This pathogen is most notable for its high gene density in its genome i.e. the genes, more than 600, encode almost 95% of the genome, which is suggestive of the fact that it has dispensed with many regulatory and biosynthetic genes for many metabolites and nutrients. By evolution, transporters have either substituted other genes or have duplicated themselves into paralogous members in Ureaplasma. SwissProt analysis and Transporter classification database were used to identify 67 open reading frames which represented the transporter dataset proteins from the whole genome. By this method, we identified a total of 20 transporters for haeme and/or iron (inclusive of binding and membrane subunits), 7 for inorganic cations (3 for Co, and one each for Mg, Cu, Ca and K), 11 for protons/ions, 5 for oligopeptides, 2 for amino acids, 2 for ammonium generated by urea hydrolysis, 4 for spermidine-putrescine, 4 for inorganic phosphate, 4 for sugars, and 2 belonging to the MOP (Multidrug-Oligosaccharidyl-lipid-Polysaccharide Flippase Superfamily) MATE (Mullti drug and Toxin Extrusion) class of membrane proteins. In addition, there are 6 other transporters, among which 5 belong to the ABC transporter family which translocate unidentified substrates. Two groups of paralogous iron transporters were identified by phylogenetic analysis using PHYLIP. Conserved residues and domains in individual transporter families were obtained by using Pro Dom and Pfam databases. In order to incorporate the missing transporters of in our work, we have also included the studies relating to the possibility of finding transmembrane-signature bearing proteins from among the hypothetical proteins. With this end in sight, we have utilized 5 different tools (TMHMM, SOSUI, HMMTOP, TopPred, and waveTM) to estimate the probable transporter proteins. Each of these tools follows completely different algorithms. Since these tools are prone to errors mainly as a result of false predictions of signal peptides as transmembrane segments, we have utilized an additional tool: SignalP, which provides to exclude the signal peptide-bearing proteins from the dataset. Transportome based studies of mucosal pathogens can give rise to many new drug and vaccine targets for prophylactic and therapeutic interventions.
Citation
The Transportome of the Human Mucosal Pathogen <i>Ureaplasma urealyticum</i> . Sudhakar, Padhmanand , Subramani, Prasanth . Lethbridge Undergraduate Research Journal. Volume 2 Number 1. 2007.
Introduction And Background
U.urealyticum is a human mucosal pathogen which derives almost all its energy from hydrolysis of urea. A member of the Mollicutes (mycoplasma) class, it is among the smallest-known free-living microorganisms [1, 64]. It belongs to the family of Mycoplasmas and is noteworthy because of the small size of its cells (0.2 to 0.3 micrometers) and of its genome (.75 Mb) [2]. This feature is largely responsible for its biologic properties, including lack of a Gram stain reaction and nonsusceptibility to many commonly prescribed antimicrobial agents, including beta-lactams. It belongs to the family of organisms which are usually associated with mucosae and mucosal membranes [64]. They reside extracellularly in the respiratory and urogenital tracts and rarely penetrate the submucosa, except in the case of immunosuppression or instrumentation, when they may invade the bloodstream and disseminate to numerous organs and tissues [60]. Ureaplasma species are the most common nonbacterial etiologies of infectious arthritis in persons who are hypogammaglobulinemic [2, 58, 60]. In several large studies, chronic lung disease of prematurity or bronchopulmonary dysplasia has also been associated with the presence of Ureaplasma organisms in the lower respiratory tract, presumably because of low-grade inflammation in the airways that causes a prolonged need for supplemental oxygen coupled with barotrauma of mechanical ventilation and oxidant damage due to oxygen administration [60]. It has also been identified as one of the causes of intrauterine-mediated diseases in newborns [60].
Reductive evolution of the genome seems to have occurred several times and to have been very rapid [3, 4, 65]. Because of its reduced genome size, and also its clinical [5] and veterinary importance, it has received much attention from sequencing centers. The genome of U. urealyticum serovar 3 is a circular chromosome comprising 751,719 bp. This is smaller than any other sequenced microbial genome except M. genitalium [6]. The G+C content is 25.5%. The genome contains 614 predicted protein-coding genes and 39 genes that code for RNAs. The genes comprise 93% of the genome. 53% of the protein-coding genes have been assigned biological roles. 19% of the genes are similar to hypothetical genes of unknown function, and 28% are unique hypothetical genes with no significant similarities to putative or demonstrated genes in other organisms [3, 4 ,5, 6].
The reduction in size of the Ureaplasma genome is thought to be related to its lifestyle, in close contact with their host [3, 6, 60, 65]. Ureaplasmas are either commensals or pathogens, and sometimes are facultative intracellular parasites [3, 4, 5]. The adoption of a parasitic life style, based on harnessing the resources of the host cell, means that numerous metabolic functions are no longer needed [58, 65]. It synthesizes indeed few precursors de novo, and the loss of genes for numerous biosynthetic pathways has been noted in the genomes of the mycoplasmas that have been sequenced to date. Ureaplasmal infections have been proved to have serious metabolic consequences. The loss of the cell wall may also be related to intracellular endoparasitism [58].
There are no genes for the de novo biosynthesis of purines or pyrimidines in U.urealyticum. Pathways for synthesis of RNA and DNA precursors are absent as is evidently shown by several notable missing enzymes [65, 66]. For example, there is no recognizable ribonucleoside-diphosphate reductase for conversion of ribonucleosides to deoxyribonucleosides, unlike in M. genitalium and M. pneumoniae [59, 64]. So U.urealyticum must import all its deoxyribonucleosides and/or deoxyribonucleoside precursors, or have a mechanism for converting ribonucleosides to deoxyribonucleosides [23, 25, 26]. As a result of its very limited biosynthetic capacity, ureaplasmas must import more of the nutrients needed for growth than do most other bacteria [26, 58, 65]. 30 different U.urealyticum transporters have been identified, which represent many transporter families. Despite this diversity of transport systems, there are a number of surprising absences, such as the previously mentioned lack of transporters for bases, nucleotides, nickel and urea [58, 65]. Missing transporters may be found among the hypothetical U.urealyticum proteins that have 5 or more predicted transmembrane regions. We have utilized five different tools to predict the proteins with transmembrane segments and domains. In this paper, we follow an approach aimed at identifying the genes and/or gene products involved in transporter-related functions in U.urealyticum. The Transport Classification (TC) system [7] represents a systematic approach to organize transport systems according to the mode of transport, energy-coupling mechanism [29], molecular phylogeny and substrate specificity. Based on TC system, Transport database- a relational database is designed for describing the cellular membrane transport proteins in organisms whose complete genome sequences are available [8]. Transport systems allow the uptake of essential nutrients and ions, excretion of end products of metabolism and deleterious substances, and communication between cells and the environment [9, 25, 58]. They also provide essential constituents of energy-generating and energy-consuming systems [10, 23, 26, 29,62]. Primary active transporters drive solute accumulation or extrusion by using ATP hydrolysis, photon absorption, electron flow, substrate decarboxylation, or methyl transfer [11, 25, 26, 62]. If charged molecules are unidirectionally pumped as a consequence of the consumption of a primary cellular energy source, electrochemical potentials result [10, 62, 68]. The consequential chemiosmotic energy generated can then be used to drive the active transport of additional solutes via secondary carriers which merely facilitate the transport of one or more molecular species across the membrane [12, 13, 25].
Methods
Identification and Classification of Transporter proteins
The completed genome and protein table for U.urealyticum strain were accessed from the NCBI ftp site (ftp://ftp.ncbi.nlm.nih.gov/genomes/). We followed the approach of Kiranmayi and Mohan, in which a stepwise procedure was used to screen out targets. All the ORFs identified in the U.urealyticum genome were BLAST searched against the Swiss-Prot/TrEMBL database and only functionally characterized proteins were taken into account. All the transporter protein coding genes were classified according to the Transporter Database (http://www.membranetransport.org/). The transmembrane domains were obtained using TOPPRED (http:// bioweb.pasteur.fr/seqanal/interfaces/toppred.html) software which is based on the Kyte-Doolittle scale of hydrophobicity [14] and Positive-inside rule. The roles played by these transporters in various biochemical and metabolic pathways were confirmed by checking for their presence in the U.urealyticum pathways registered in the Kyoto Encyclopedia of Genes and Genomes Database (KEGG) (http://www.genome.jp/kegg/pathway.html). Domain searches for the transporter proteins were carried out using the Pro Dom server (http://prodom.prabi.fr/prodom/current/html/home.php) and Pfam (http://www.sanger.ac.uk/Software/Pfam/). Phylogenetic analysis was performed using TREETOP software (http://www.genebee.msu.su/services/phtree_reduced.html) and trees were bootstrapped 100 times to ensure the reliability of each branch point [21].
Analyses of Hypothetical proteins
The hypothetical proteins, inclusive of unique putative membrane lipoproteins and conserved hypothetical proteins from the U.urealyticum genome were analyzed for the presence of transmembrane signatures using online tools like HMMTOP (http://www.enzim.hu/hmmtop/) [15], SOSOI (http://sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html) [16], TopPred II (http://www.sbc.su.se/~erikw/toppred2/), [17] and TMHMM (http://www.cbs.dtu.dk/services/TMHMM-2.0/) [18] available through the Expasy server. In addition, another tool called wave TM (http://athina.biol.uoa.gr/bioinformatics/waveTM/) [19] was also used for the prediction of transmembrane signatures. Proteins predicted to contain transmembrane segments by TMHMM, HMMTOP, SOSUI, TopPred and WaveTM were tested further using SignalP version 3.0 (http://www.cbs.dtu.dk/services/SignalP/), [20]. Default conditions were used for the predictive analyses. A positive prediction of signal peptides by SignalP was considered to start within the first 70 amino acids of the N-termini of the proteins.
Results and Discussion
Characterizing the Transporter Dataset
We have identified 67 (inclusive of subunits of individual transporters) transporters by integrated computer based analysis of the Ureaplasma proteins. These transporters fall into various categories involved in the transport of many metabolites and nutrients, among which some of them are those transporting aminoacids, polypeptides, deoxyribonucleosides and/or deoxyribonucleoside precursors, and inorganic metal ions of cobalt, copper, iron, potassium, and magnesium which function as cofactors for many enzymes [FIGURE 1]. The transporters are placed in their respective families according to the TC system of classification based on characteristic features like transmembrane domains, hydropathic plots, conserved motifs/ residues and molecular phylogeny. All the 67 transporter protein-related ORFs which have been identified are summarized in TABLE 1. The homologs of the Ureaplasma transporters in other prominent bacterial pathogens are summarized in TABLE 2.
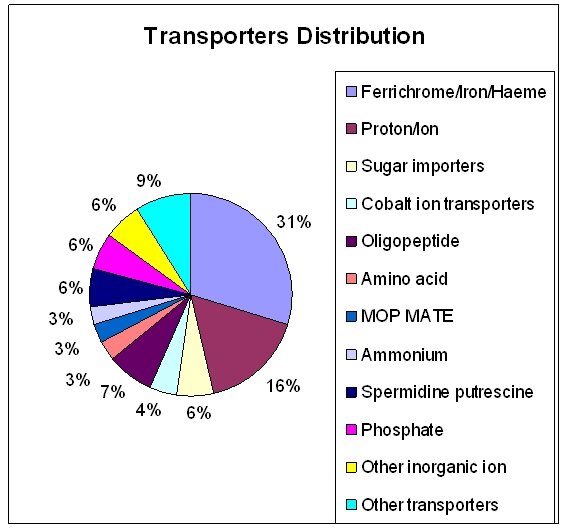
Figure 1: Schematic representation of the Ureaplasma urealyticum transporter protein classification
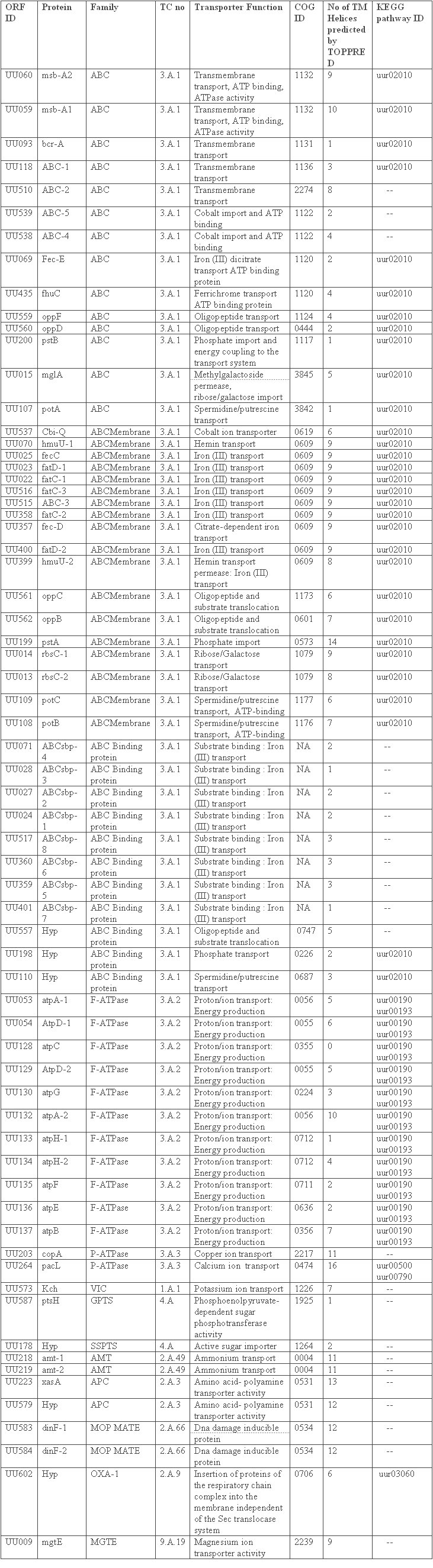
Table 1: Transporter genes of Ureaplasma urealyticum. ABC: ATP Binding Cassette, VIC: Voltage Gated Ion Channel, GPTS: General Phosphotransferase System, SSPTS: Sugar Specific Phosphotransferase System, AMT: Ammonium Transporter, APC: Amino acid – Polyamine Organocation, MOP: Multidrug/oligosaccharidyl-lipid/ Polysaccharide, OXA: Cytochrome Oxidase Biogenesis, MGTE: Mg 2+ ion Transporter E.
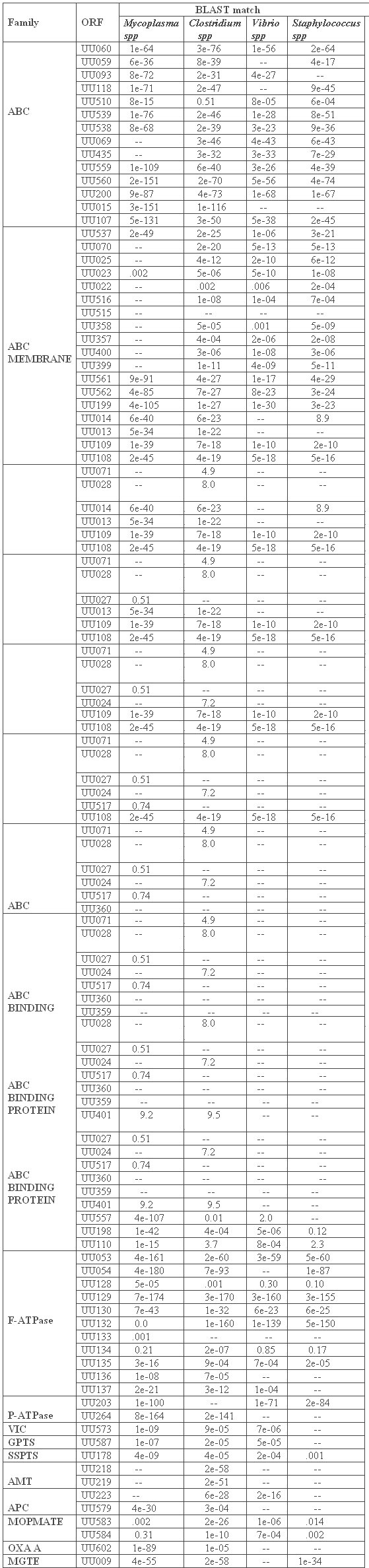
Table 2: Homology levels of the Ureaplasma urealyticum transporters with respect to their counterparts in prominent bacterial pathogens. The values indicate how much the transporter proteins have diverged from or converged towards the representative species
F-ATPase Enzyme Complex
U.urealyticum lacks the conventional mechanisms for adenosine 5-triphosphate (ATP) generation, such as glycolysis or arginine breakdown, present in other mycoplasmas. The energy yielding ATP molecules are generated through the formation of an ion gradient coupled to urea hydrolysis [23, 26, 67]. U. urealyticum contains an enzyme complex involved in energy production via proton efflux. This family of enzymes, belonging to the functional class of energy metabolism and ATP-proton motive force interconversion comprises two oligomeric complexes: F1 - the catalytic core and FO - the membrane proton channel. F1 has five subunits: Alpha(3), Beta(3), Gamma(1), Delta(1) and Epsilon(1) [62, 67]. FO has three main subunits: A, B and C [22, 26, 67]. The ureaplasma subunit of the F1 complex, which in other organisms is the interface between the FO and F1 subunits and causes the permeability of the transmembrane proton channel FO, is present as two distinct peptides [67]. UU134 corresponds to the two-thirds of the amino-terminal of the eubacterial consensus, and UU133 corresponds to the normally more conserved carboxyl terminus. This non-paralogous gene cluster (UU053, UU054, UU128-UU130, and UU132-UU137) is a likely candidate for the ATP synthase activity seen in conjunction with the urease present in Ureaplasma urealyticum. Speculatively, these atypical features may be linked to the unique ATP production pathways of ureaplasmas [62]. The source for 5% of ureaplasma ATP production resulting from urea hydrolysis are most probably from substrate phosphorylation [22]. Enzymatic studies show that U. urealyticum has a limited capacity to metabolize carbohydrates [22, 23, 26].
Bacterial F-type ATPases pump 3-4 H + and Na + out of the cell per ATP hydrolyzed [67]. These enzymes also operate in the opposite direction, synthesizing ATP when protons flow through the 'ATP synthase' down the proton electrochemical gradient (the 'proton motive force' or pmf) [26, 68]. V-type ATPases may pump 2-3 H + per ATP hydrolyzed, and these enzymes cannot catalyze pmf-driven ATP synthesis. It has been proposed that this difference between F-type and V-type ATPases is due to a 'proton slip' that results from an altered structure in the membrane sector of V-type ATPases [68]. It has also been shown that the Ureaplasma's membrane bound ATPase enzyme complex has similarities to the FO F1 ATPases of other bacteria [23, 25, 62, 67]. It has also been reported that ATP determination represents a reliable and accurate means of measuring growth [24]. It is therefore attractive to propose that an FO F1 membrane ATPase in U.urealyticum plays an important role in energy generation [62]. This could in turn rely upon a chemiosmotic mechanism [25, 26], whereby generation of a transmembrane electrochemical potential would provide the electrochemical force for protons to enter the cell via the FO F1 membrane ATPase to generate ATP [67]. Such electrochemical potentials may also be used in transport processes. It has been suggested that since U.urealyticum has a potent cytosolic urease, urea hydrolysis to ammonium ions and carbon dioxide generates a transmembrane potential which drives ATP synthesis [26, 58, 61, 69]. It is worth mentioning that the pH of the urogenital tract is usually on the acidic side of neutrality, corresponding to the pH values optimal for ureaplasma growth, maximum increase in delta P, maximum ammonia chemical potential, maximum urease activity, and maximum ATP generation [25, 26]. Hence targeting the components of this indispensable energy generating system of Ureaplasma would be a good option to block growth and multiplication.
Iron transporters
Unexpected among the 19 ABC transporters are many different Fe 3+ and/or haemin transporters. Iron is essential for the function of many enzymes [29]. However, the low solubility of Fe 3+ at physiological pH forces Ureaplasma urealyticum to develop special mechanisms to acquire necessary amounts. Phylogenetic analysis of the ten membrane-spanning protein subunits (UU070, UU025, UU023, UU022, UU516, UU515, UU358, UU357, UU399, and UU400) of the U. urealyticum iron transporters indicated that they all cluster together with respect to other known bacterial proteins [FIGURE 2] and form two related, but distinct paralogous groups within this U. urealyticum gene family [FIGURE 3]. Except for UU399 which has 8 membrane spanners, all the other iron transporters are characterized by the presence of 9 transmembrane segments in the N-terminus. This paralogous family arose from gene duplication events during ureaplasma evolution [70, 71]. In the apparent absence of the complement of transcriptional regulators found in most other bacteria, it is speculated that U. urealyticum increased its capacity to import iron by increasing the number of iron transporter genes [70]. This indicates that the availability of iron may be a limiting factor of ureaplasma growth [29, 71]. Iron is associated with the membranes of Mycoplasma capricolum [ 27 ] and M. pneumoniae [28]. In these mycoplasmas, the iron may act as a reversible electron carrier in concert with NADH oxidase, or as a component of an iron-containing electron storage protein [28]. Unlike the other mycoplasmas, U. urealyticum lacks both NADH oxidase activity [29] and an NADH oxidase gene. Even if ureaplasmas use iron in their respiration, the iron use is probably different from that in other mycoplasmas [27, 28, 70].
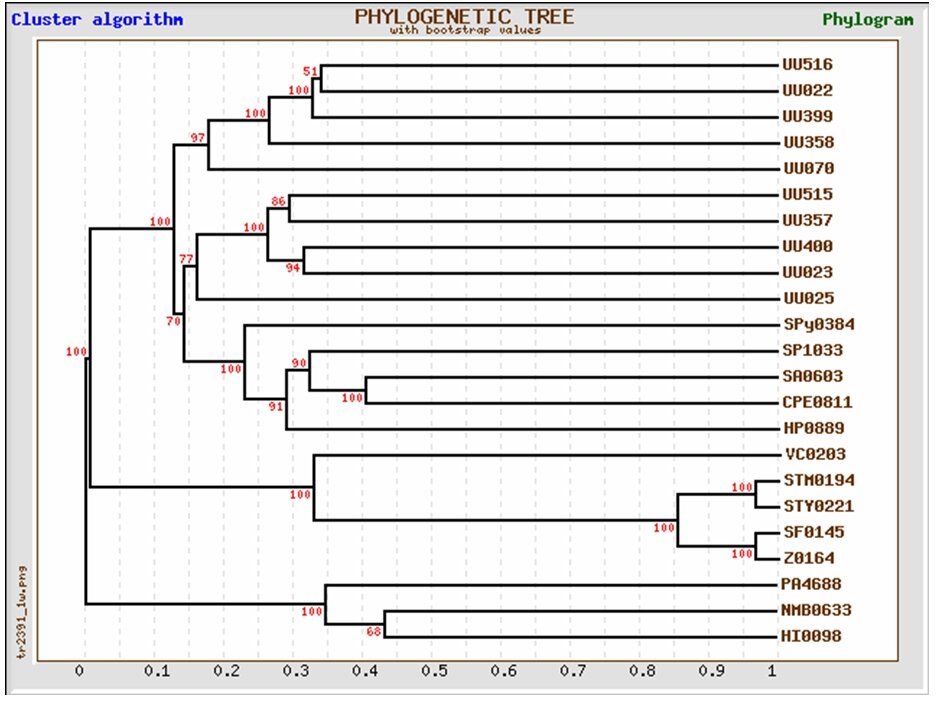
Figure 2: Phylogeny of the membrane spanning subunits of the ten iron transporters of Ureaplasma urealyticum with respect to iron transporting genes of other pathogens

Figure 3: Phylogenetic representation of the paralogous nature of the iron transporters in Ureaplasma urealyticum. The tree shows the two paralogous groups of iron transporters, most of which are copies of one essential parent iron-transporter gene
MOP MATE Family
The MOP flippase superfamily includes four distantly related families, among which is the ubiquitous family (MATE) specific for drugs. All functionally characterized members of the MOP superfamily catalyze efflux of their substrates, presumably by cation antiport [78]. The MATE family includes a functionally characterized multidrug efflux system from Vibrio parahaemolyticus NorM [30, 31, 78], and several homologues from other closely related bacteria (including U.urealyticum) that function by a drug:Na + antiport mechanism. Some of the other members of this family include a putative methionine resistance protein of Saccharomyces cerevisiae, a cationic drug efflux pump in A. thaliana [32] and the functionally uncharacterized DNA damage-inducible protein F (DinF) of E. coli [33, 34, 78]. In Ureaplasma, there are two paralogous proteins (UU583 and UU584) of this family. These are of about 450 amino acyl residues in length and exhibit 12 putative TMS. They arose by an internal gene duplication event from a primordial 6 TMS encoding genetic element. The yeast proteins are larger (upto about 700 residues) and exhibit about 12 TMSs. The family includes hundreds of functionally uncharacterized but sequenced homologues from bacteria, archaea, and all eukaryotic kingdoms. The members of this family in Ureaplasma can be used as very efficient and effective targets in co-drug therapies. Protein domain searches show that residues 165-298 are 26% similar to a defined domain of O34474_BACSU, a virulence factor.
Ammonium Transporters
U. urealyticum generates 95% of its ATP through the hydrolysis of urea by urease [22]. Since the discovery in 1966 that ureaplasmas metabolize urea, the physiology of this energetic process has been studied to gain an understanding of what appears to be a unique system [35, 61]. The hypothesis is that hydrolysis of urea generates an electrochemical gradient through the accumulation of intracellular ammonia/ammonium [61]. This gradient fosters a chemiosmotic potential that generates ATP. The intracellular concentration of ammonia generated through urea hydrolysis in U. urealyticum has been measured to be 21 times the extracellular concentration [63]. This excess suggests efflux of ammonium/ammonia through a saturable uniporter rather than by passive diffusion [22, 61, 63]. A pair of paralogous genes that code for ammonia transporters of the Amt family were identified (UU218, UU219). Pfam searches place residues 48 to 463 of UU218 in the Ammonium Transporter Family (PF00909) [63]. Homologs occur in Gram-negative and Gram-positive bacteria, archaea, yeast, plants and animals and are probably ubiquitous. Although the physiology of Amt family transporters is controversial [36, 37, 38, 63], the probable mechanism in ureaplasma is energy-independent-facilitated diffusion [35, 61, 62]. The electrochemical gradient created by the export of ammonium presumably results in an ATP molecule generating proton influx through the U. urealyticum F-type ATPase. The proteins of the Amt family vary in size from 391 to 622 amino acyl residues and possess 11 (N-terminus out; most members) or 12 (N-terminus in) transmembrane α-helical spanners. Because of their channel-like properties, these proteins might more appropriately be classified in TC class 1.A. However, because of several reports suggesting that some members exhibit carrier properties, the family is retained in class 2.A for the present.
P-ATPase type cation transporters
Nearly all of the members of this superfamily of P-ATPases, found in bacteria, archaea and eukaryotes, catalyze cation uptake and/or efflux driven by ATP hydrolysis [79]. Most of these protein complexes are multisubunits with a large subunit serving the primary ATPase and ion translocation functions. The Ca2+ ATPase (UU264) reported as a paralog to UU203, catalyzes calcium ion antiport. A single organism may possess multiple isoforms of this protein entity [79]. Some members of the P-type ATPase family have been reported to flip phospholipids from one monolayer of the bilayer membrane to the other monolayer [39, 40, 79]. Protein Domain searches of the calcium ion transporter reveal that residues 8-179 are 36% similar to a defined domain of ATC_ARTSF, residues 262-353 are 55% similar to O59868_SCHPO, and residues 474-557 are 52% similar to ATCL_MYCGE, all of which are described as ATPase proteins. It is also characterized by the presence of about 16 transmembrane helices from the N- terminal region. The paralog, UU203, a copper ion transporting ATPase belongs to the functional class of cation transport and binding proteins [39]. As this sequence contains similarity to ATPases, and contains 11 transmembrane regions, it is possible that this represents an export protein [40]. Curiously, the closest domain comes from a yeast sequence: residues 233-463 are 33% similar to residues 478-717 of ATU2_YEAST, a predicted P-type copper-transporting ATPase.
VIC Potassium Channel Protein
The K + channels usually consist of homotetrameric structures with each α-subunit possessing six transmembrane spanners (TMSs). Many voltage-sensitive K + channels function with β-subunits that modify K + channel gating [41]. These nonintegral β-subunits are oxidoreductases that co-assemble with the tetrameric α-subunits in the endoplasmic reticulum and remain tightly adherent to the α-subunit tetramer. The high resolution β-subunit structure is available [41]. Evolutionary relationships between K + channels and certain K + :cation symporters has been reviewed and discussed [42]. It has been shown to form tetramers that may function to maintain the membrane potential in the early stationary phase of growth [43]. No paralogs have been found and protein domain searches show that residues 219-318 are 30% similar to a defined domain of O67715_AQUAE, an ion channel.
The Amino Acid-Polyamine-Organocation transporters (APC)
The APC superfamily of transport proteins includes members that function as solute:cation symporters and solute:solute antiporters [44]. They occur in bacteria, archaea, yeast, fungi, unicellular eukaryotic protists, slime molds, plants and animals [44]. They vary in length, being as small as 350 residues and as large as 850 residues. Most of them possess twelve transmembrane α-helical spanners but have a re-entrant loop involving TMSs 2 and 3 [45]. The 2 representatives (UU223 and UU579) of this family in U.urealyticum are non-paralogous. Residues 28-100 of UU223 constitute a domain of XASA_ECOLI, a transmembrane amino acid permease.
GPTS sugar transporter
The Phosphoenolpyruvate-dependent Sugar PhosphoTransferase system (PTS) is a major carbohydrate transport system in Ureaplasma. The PTS catalyzes the phosphorylation of incoming sugar substrates concomitant with their translocation across the cell membrane. The general mechanism of the PTS is as follows: a phosphoryl group from phosphoenolpyruvate (PEP) is transferred to Enzyme I (EI) of the PTS which in turn transfers it to a phosphoryl carrier protein (HPr) [46, 72, 73]. Phospho-HPr then transfers the phosphoryl group to a sugar-specific permease complex (enzymes EII/EIII) [46, 73]. UU587, a phosphohistidinoprotein-hexose phosphotransferase, is the representative of this family of sugar transporters in U.urealyticum and thus plays a significant role in acquiring sugars required for growth and energy from the host [72, 74]. Residues 1 to 84 place UU587 in the PTS-HPr family which is described as PTS HPr component phosphorylation site (PF00381) according to Pfam.
SSPTS sugar (glucose) transporter
UU178 is another type of non paralogous transporter of sugars, especially glucose. SSPTS, as the name suggests, is a sugar specific transporter family found in many bacteria [73]. This Glc family includes porters specific for glucose, glucosamine, N-acetylglucosamine and a large variety of α- and β-glucosides. However, not all β-glucoside PTS porters are in this class, as the PTS porter first described as the cellobiose (Cel) β-glucoside porter is the diacetylchitobiose porter in the Lac family (TC #4.A.3). The IIA, IIB and IIC domains of all of the group translocators are demonstrably homologous [73, 75]. Several of the PTS porters in the Glc family lack their own IIA domains and instead use the glucose IIA protein (IIA glc or Crr). Most of these porters have the B and C domains linked together in a single polypeptide chain. A cysteyl residue in the IIB domain is phosphorylated by direct phosphoryl transfer from IIA glc (his~P) or one of its homologues [77]. Those porters which lack the IIA domain include the maltose (Mal), arbutin-salicin-cellobiose (ASC), trehalose (Tre), putative glucoside (Glv) and sucrose (Scr) porters of E. coli [74, 75]. Most, but not all Scr porters of other bacteria also lack a IIA domain. Residues 12-123 are 35% similar to a defined domain of Y129_MYCGE [76, 77].
The Magnesium ion transporter
Belonging to the functional class of cation transport and binding proteins, this ubiquitous transporter protein (UU009) has many sequenced homologs in a wide variety of bacteria, archaea and eukaryotes. Its phylogeny roughly follows that of the organismal taxonomies [48]. The homologs have sizes that vary considerably from 311 residues for the Methanococcus thermoautotrophicum protein, 463 residues for the Synechocystis homologue, and 513 residues for the human homolog, SLC41A1. The B. firmus transporter and several homologs examined have strongly charged, hydrophilic N-terminal domains (cytoplasmic) followed by a hydrophobic C-terminal domain with 5 putative transmembrane α-helical spanners [48]. The central 100 residues resembles archaeal inositol monophosphate dehydrogenases [47]. The modes of transport, the nature of the energy source and the potential reversibility of the transport reaction have not been studied. However, the available evidence suggests that the MgtE proteins are secondary carriers with inwardly directed polarity [47, 48]. Residues 146-263 are 28% similar to an inosine-5'-monophosphate CBS domain as seen in YC32_METJA.
Prediction of Transmembrane Segments in Hypothetical Proteins
For the identification of transmembrane signatures in the hypothetical proteins of U.urealyticum, we have used a range of computational tools. The most primitive tools relied on the hydrophobicity of transmembrane helices [14]. The continuing development of additional strategies has improved their predictive accuracies. Some, for example, examine the whole amino acid sequence of a membrane protein [49, 50]. Such tools also search for the distribution of particular amino acids since, in general, the cytoplasmic side of integral membrane proteins is positively charged, a phenomenon known as the “positive-inside rule” [17]. A substantial improvement has been achieved with the utilization of hidden Markov model (HMM) algorithms [15]. However, a major pitfall of all methods is the erroneous prediction of the N-terminal signal peptides as transmembrane segments as a result of their hydrophobic nature. We utilized five different computational tools that are based on four different algorithm approaches in order to predict integral membrane proteins in the Ureaplasma proteome. The tools were: TopPred II, which scans proteins for hydrophobic stretches of amino acids and the inside-positive phenomenon; SOSUI, which examines a combination of amino acid hydrophobicity, amphiphilicity of the transmembrane segment, amino acid charges, and length of transmembrane segment; and TMHMM and HMMTOP, both of which are based on hidden Markov model algorithm. A newer method for predicting transmembrane segments, waveTM, was also utilized. WaveTM uses a mathematical function known as wavelet, which measures the hydrophobicity of a sliding 20-amino acid window.
A total of 283 proteins of unknown function from the Ureaplasma proteome were subjected to this study. It was thought that the missing transporters can be found among the uncharacterized hypothetical proteins. SOSUI predicted the fewest integral membrane proteins (107 proteins or 37.80% of total protein set of 283); TopPred II predicted the largest number of proteins to contain membrane spanning segments (245 proteins or 86.57%). The other three programs estimated integral membrane proteins within this range with TMHMM predicting 112 proteins (39.57%), HMMTOP predicting 137 proteins (48.40%), and waveTM predicting 119 proteins (42.04%)[TABLE 3]. Almost half of the proteins classified as integral membrane proteins by TMHMM, SOSUI, HMMTOP and waveTM were predicted to contain a single transmembrane segment (53.37%, 48.59%, 50.36% and 51.26 % respectively). But TopPred II predicted the fewest proteins with one transmembrane segment (29.38%). Prediction of proteins with 5 or more transmembrane segments was more coherent [TABLE 3]. The range of positive predictions for transmembrane proteins with 5 or more TMSs falls between 16.05% (HMMTOP) and 21.49% (SOSUI).
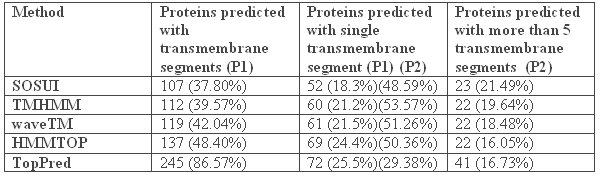
Table 3: Analysis of the hypothetical proteins of Ureaplasma urealyticum using various tools to detect the presence of transmembrane signatures. P1 – Ratio based on the total number of hypothetical proteins taken for analysis (283). P2 – Ratio is calculated on the basis of the number of proteins predicted with transmembrane segments for each tool.
In order to eliminate false-positive predictions, a sixth method, SignalP, which discriminates between signal peptides and transmembrane segments, has also been utilized. Due to the hydrophobic nature of signal peptides, all programs used in this study are susceptible for erroneous classification of signal peptides as transmembrane segments. Therefore, we tested for possible false-positive predictions among the proteins predicted to contain transmembrane segments using SignalP. This tool combines two computational methods, namely neural networks and hidden Markov model, to predict the presence of signal peptides based on amino acid composition and cleavage site of signal peptides. After removing the false positives by SignalP, we can conclude that TMHMM has predicted 73 proteins, SOSUI 88 proteins, HMMTOP 115 proteins, TopPred 235 proteins and waveTM 89 proteins as positive predictions or in other words hypothetical proteins which have been predicted to be non-secretory and hence integral to the membrane. These predictions constituted 25.79%, 31.09%, 40.63%, 83.03% and 31.44% of all the hypothetical proteins tested (283), respectively [TABLE 4].
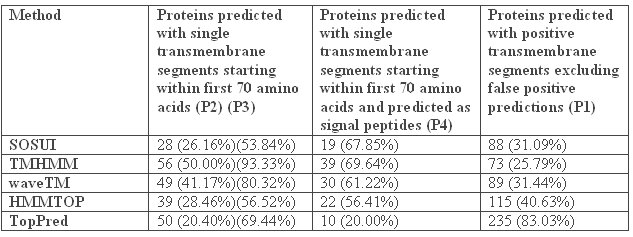
Table 4: Final prediction of non-secretory transmembrane proteins among the hypothetical proteins in Ureaplasma urealyticum using SignalP. P1 – Ratio based on the total number of hypothetical proteins taken for analysis (283). P2 – Ratio is calculated on the basis of the number of proteins predicted with transmembrane segments for each tool. P3 – Ratio is calculated on the basis of the number of proteins predicted with single transmembrane segment for each tool. P4 – Ratio is calculated on the basis of the number of proteins predicted with single transmembrane segments in the first 70 amino acids.
Membrane transporters as drug targets
Comparative analysis of complete genomes revealed that most of the pathogens have drastically diminished biosynthetic capabilities as compared to their free-living relatives [51, 52]. Instead, these organisms depend on their hosts to provide essential nutrients such as amino acids, nucleobases, and vitamins. Transport systems for these nutrients are generally well conserved and easily identifiable [53]. Analysis of metabolic pathways allows one to predict which substrates cannot be produced inside the cells and so need to be transported [54]. All this makes bacterial transport proteins attractive drug targets. A number of non-metabolizable analogs of amino acids and nucleobases are already available and can be easily checked for their potential antibacterial activity. Finally, an interesting approach that has emerged only recently includes improving activity of the existing antibiotics by inhibiting bacterial multidrug transporters [52]. Genomes of many pathogenic bacteria appear to contain homologs of multidrug pumps, such as MOPMATE protein in U.urealyticum, which protects bacterial cells by exporting antibiotic molecules. Inhibiting such pumps not only creates convenient model organisms for studying drug effectiveness [56] but also allows resistance to classical drugs such as tetracycline to be overcome [57]. The nearly universal distribution of most major classes of transport proteins [55], however, makes it probable that effective inhibitors of bacterial transport turn out to be also toxic for humans. So we must sort out those essential transporters which have no significant homologs in humans as the most probable and suitable drug targets. Extracellular subunits of these transporters can also be used as vaccine targets.
Conclusion
Mucosal pathogens like those in the mycoplasmal family predominantly rely on the host for nutrient supply as they have lost many of the genes or genetic clusters coding for biosynthetic pathways, thus making these pathways incomplete or obsolete. As a result, transporter proteins are so critical for the pathogens' survival and multiplication. The availability of the genome sequence of U.urealyticum has helped in the primary identification of transporter genes. Proteomics also holds good potential to achieve biomedical milestones in many disciplines, in particular the discovery of drug targets. Choosing an optimal target function is a crucial step in the long and expensive process of drug development and requires the best possible understanding of related biological processes in bacterial pathogens and their hosts. A class of proteins of special interest is transmembrane proteins. The significance of these proteins is that they constitute more than 45% of current drug targets. Ureaplasma has been found to encode more transporters than other members of the mycoplasma family. The presence of two paralogous groups of iron transporters in Ureaplasma suggests that gene duplication has occurred and that iron is very essential for so many activities of this deadly emerging pathogen [70]. Ammonium transporters are yet another class of transporters which are very essential to Ureaplasma as they are involved in the creation of the electrochemical gradient by the export of ammonia, which therefore results in ATP generation. In addition, there is an array of transporters ranging from the ABC subtypes involved in cation transport to the drug efflux pumps. Advanced biochemical characterization and laboratory studies will give rise to much important information about the efficacy of using these transporter proteins as drug targets.
Endnotes
1. Woese CR; Stackebrandt E; Ludwig W. What are mycoplasmas: the relationship of tempo and mode in bacterial evolution. J Mol Evol 21: 305-16 (1984).
2. Borkhsenius SN; Chernova OA. The Mycoplasma genome. Tsitologiia 29: 619-31 (1987).
3. Sladek TL. A hypothesis for the mechanism of mycoplasma evolution. J Theor Biol 120: 457-65 (1986).
4. Syvanen M. Molecular clocks and evolutionary relationships: possible distortions due to horizontal gene flow. J Mol Evol 26: 16-23 (1987).
5. Brown RB. Community-acquired bronchitis and pneumonia. Presentation, pathogens, and therapy. Postgrad Med 79: 241-50 (1986).
6. Fraser, C. M. et al. The minimal gene complement of Mycoplasma genitalium. Science 270, 397-403 (1995).
7. Saier, M. H., jr.. A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol. Mol. Biol. Rev. 64, 354-411 (2000).
8. Ren, Q., Kang, K. H. and Paulsen, I. T. (2004). TransportDB: a relational database of cellular membrane transport systems. Nucleic Acids. Res. 32, D284-D288 (2004).
9. Mitchell P. Translocations through natural membranes. Adv Enzymol. ;29:33-87 (1967).
10. Mitchell P. Proton-translocation phosphorylation in mitochondria, chloroplasts and bacteria: natural fuel cells and solar cells. Fed Proc.;26:1370-1379 (1967).
11. Dimroth P. Primary sodium ion translocating enzymes. Biochim Biophys Acta.1318:11-51 (1997).
12. Maloney P C. Microbes and membrane biology. FEMS Microbiol Rev. ;87:91-102 (1990).
13. Maloney P C. The molecular and cell biology of anion transport by bacteria. Bioessays ;14:757-762 (1992).
14. Kyte, J. and Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105-132 (1982)..
15. Tusnády, G. E. and Simon, I.. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283, 489-506 (1998).
16. Hirokawa, T., Boon-Chieng, S. and Mitaku, S. SOSUI: classification and secondary structure prediction system for membrane protein. Bioinformatics 14, 378-379(1998).
17. Von Heijne, G.. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487-494 (1992).
18. Krogh, A., Larsson, B., von Heijne, G. and Sonnhammer, E. L. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genome., J. Mol. Biol. 305, 567-580 (2001)..
19. Pashou, E. E., Litou, Z. I., Liakopoulos, T. D. and Hamodrakas, S. J.. waveTM: Wavelet-based transmembrane segment prediction. In Silico Biol. 4, 0012 (2004).
20. Bendtsen, J. D., Jensen, L. J., Blom, N., von Heijne, G. and Brunak, S. Feature based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 17, 349-356 (2004)..
21. Yushmanov, S. V. and Chumakov, K. M. Algorithms of the maximum topological similarity phylogenetic trees construction. Mol. Gen. Microbiol. Virusol. 3, 9-15 (1988).
22. Smith, D. G. , Russell, W. C. , Ingledew, W. J. & Thirkell, D. Hydrolysis of urea by Ureaplasma urealyticum generates a transmembrane potential with resultant ATP synthesis. J. Bacteriol. 175, 3253-3258 (1993).
23. Romano, N., G. Tolone., and R. La Licata.. Adenosine triphosphatase activity of Ureaplasma urealyticum. Microbiologica 5: 25-33 (1982).
24. Stemler, M.E., G.W.Stemke., and J.A. Robertson. ATP measurements obtained by luminometry provide rapid estimation of Ureaplasma urealyticum growth. J.Clin.Microbiol. 25: 427-429 (1987).
25. Mitchell.P.,. Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature: 191: 144-148 (1961).
26. Masover, G.K., S.Razin, and L.Hayflick.. Localization of enzymes in Ureaplasma urealyticum. J.Bacteriol. 130: 297-302 (1977).
27. Bauminger, E. R. et al. Iron storage in Mycoplasma capricolum. J. Bacteriol. 141, 378-381 (1980).
28. Pollack, J. D. , Merola, A. J. , Platz, M. & Booth, R. L. J. Respiration-associated components of Mollicutes. J. Bacteriol. 146, 907-913 (1981).
29. Pollack, J. D. , Williams, M. V. & McElhaney, R. N. The comparative metabolism of the mollicutes (Mycoplasmas): the utility for taxonomic classification and the relationship of putative gene annotation and phylogeny to enzymatic function in the smallest free-living cells. Crit. Rev. Microbiol. 23, 269- 354 (1997).
30. Brown, M.H., I.T. Paulsen, and R.A. Skurray. The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol. Microbiol. 31: 394-395 (1999).
31. Chen, J., Y. Morita, M.N. Huda, T. Kuroda, T. Mizushima, and T. Tsuchiya. VmrA, a member of a novel class of Na+-coupled multidrug efflux pumps from Vibrio parahaemolyticus. J. Bacteriol. 184: 572-576 (2002).
32. Diener, A.C., R.A. Gaxiola, and G.R. Fink. Arabidopsis ALF5, a multidrug efflux transporter gene family member, confers resistance to toxins. The Plant Cell 13: 1625-1637 (2001).
33. Morita, Y., K. Kodama, S. Shiota, T. Mine, A. Kataoka, T. Mizushima, and T. Tsuchiya. NorM, a putative multidrug efflux protein, of Vibrio parahaemolyticus and its homolog in Escherichia coli. Antimicrob. Agents Chemother. 42: 1778-1782 (1998).
34. Nishino, K. and A. Yamaguchi. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183: 5803-5812 (2001).
35. Shepard, M. C. Human mycoplasma infections. Health Laboratory Science 3, 163-169 (1966).
36. Siewe, R. M. et al. Functional and genetic characterization of the (methyl)ammonium uptake carrier of Corynebacterium glutamicum. J. Biol. Chem. 271, 5398-5403 (1996).
37. Jayakumar, A. , Epstein, W. & Barnes, E. M. J. Characterization of ammonium (methylammonium)/potassium antiport in Escherichia coli. J. Biol. Chem. 260, 7528-7532 (1985).
38. Soupene, E. , He, L. D. Y. & Kustu, S. Ammonia acquisition in enteric bacteria: physiological role of the ammonium/methylammonium transport B (AmtB) protein. Proc. Natl Acad. Sci. USA 95, 7030- 7034 (1998).
39. Kühlbrandt, W., M. Auer, and G.A. Scarborough. Structure of the P-type ATPases. Curr. Opin. Struc. Biol. 8: 510-516 (1998)..
40. Rensing, C., M. Ghosh, and B.P. Rosen. Families of soft-metal-ion transporting ATPase. J. Bacteriol. 181: 5891-5897 (1999).
41. Gulbis, J.M., S. Mann, and R. MacKinnon. Structure of a voltage-dependent K+ channel β-subunit. Cell 97: 943-952 (1999).
42. Durell, S.R., Y. Hao, T. Nakamura, E.P. Bakker, and H.R. Guy. Evolutionary relationship between K+ channels and symporters. Biophys. J. 77: 775-788 (1999).
43. Ungar, D., A. Barth, W. Haase, A. Kaunzinger, E. Lewitzki, T. Ruiz, H. Reiländer, and H. Michel. Analysis of a putative voltage-gated prokaryotic potassium channel. Eur. J. Biochem. 268: 5386-5396 (2001).
44. Saier, M.H., Jr. Families of transmembrane transporters selective for amino acids and their derivatives. Microbiology 146: 1775-1795 (2000).
45. Gasol, E., M. Jiménez-Vidal, J. Chillarón, A. Zorzano, and M. Palacín. Membrane topology of system xc - light subunit reveals a re-entrant loop with substrate-restricted accessibility. J. Biol. Chem. 279: 31228-31236 (2004).
46. Reizer, J., V. Michotey, A. Reizer, and M.H. Saier, Jr. Novel phototransferase system genes revealed by bacterial genome analysis: unique, putative fructose- and glucoside-specific systems. Prot. Sci. 3: 440-450 (1994).
47. Kehres, D.G. and M.E. Maguire. Structure, properties and regulation of magnesium transport proteins. Biometals 15: 261-270 (2002).
48. Smith, R.L., L.J. Thompson, and M.E. Maguire. Cloning and characterization of MgtE, a putative new class of Mg2+ transporter from Bacillus firmus OF4. J. Bacteriol. 177: 1233-1238 (1995).
49. Jones, D. T., Taylor, W. R. and Thornton, J. M. A mutation data matrix for transmembrane proteins. FEBS Lett. 339, 269-275 (1994).
50. Taylor, W. R., Jones, D. T. and Green, N. M. A method for alpha-helical integral membrane protein fold prediction. Proteins 18, 281-294 (1994).
51. Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA,Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM et al.:The minimal gene complement of Mycoplasma genitalium. Science 270:397-403 (1995).
52. Lewis K: Multidrug resistance: versatile drug sensors of bacterial cells. Curr Biol, 9:R403-R407 (1999).
53. Clayton RA, White O, Ketchum KA, Venter JC: The first genome from the third domain of life. Nature, 387:459-462 (1997).
54. Galperin MY, Tatusov RL, Koonin EV: Comparing microbial genomes: how the gene set determines the lifestyle. In Organization of the Prokaryotic Genome. Edited by Charlebois RL. Washington: ASM Press;:91-108 (1999).
55. Saier MH Jr: Eukaryotic transmembrane solute transport systems. Int Rev Cytol, 190:61-136 (1999).
56. Hsieh PC, Siegel SA, Rogers B, Davis D, Lewis K: Bacteria lacking a multidrug pump: a sensitive tool for drug discovery. Proc Natl Acad Sci USA, 95:6602-6606 (1998).
57. Nelson ML, Levy SB: Reversal of tetracycline resistance mediated by different bacterial tetracycline resistance determinants by an inhibitor of the Tet(B) antiport protein. Antimicrob Agents Chemother, 43:1719-1724 (1999).
58. McCormack WM. Ureaplasma urealyticum ecological niche and epidemiolocal considerations. Pediatr Infect Dis J; 5:S232-3 (1986).
59. Himmelreich, R. et al. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 24, 4420-4429 (1996).
60. Cassell, G. H. , Waites, K. B. , Watson, H. L. , Crouse, D. T. & Harasawa, R. Ureaplasma urealyticum intrauterine infection: role in prematurity and disease in newborns. Clin. Microbiol. Rev. 6, 69-87 ( 1993).
61. Mobley, H. L. , Island, M. D. & Hausinger, R. P. Molecular biology of microbial ureases. Microbiol. Rev. 59, 451-480 (1985).
62. Romano, N. , La Licata, R. & Russo Alesi, D. Energy production in Ureaplasma urealyticum. Pediatr. Infect. Dis. 5, 308- 312 (1986).
63. Soupene, E. , He, L. D. Y. & Kustu, S. Ammonia acquisition in enteric bacteria: physiological role of the ammonium/methylammonium transport B (AmtB) protein. Proc. Natl Acad. Sci. USA 95, 7030- 7034 (1998).
64. Himmelreich, R. , Plagen, H. , Hilbert, H. , Reiner, B. & Herrmann, R. Comparative analysis of the genomes of the bacteria Mycoplasma pneumoniae and Mycoplasma genitalium. Nucleic Acids Res. 25, 701-712 ( 1997).
65. Hutchison, C. A. et al. Global transposon mutagenesis and a minimal mycoplasma genome. Science 288, 2165-2169 (1999).
66. Romano N, Tolone G, La Licata R. Adenosine triphosphatase activity of Ureaplasma urealyticum. Microbiologica. ; 5(1):25-33 (1982).
67. Zilberstein D, Shirvan MH, Barile MF, Rottem S. The beta-subunit of the F1F0-ATPase is conserved in mycoplasmas. J Biol Chem.;261(16):7109-7111 (1986).
68. Benyoucef M, Rigaud JL, Leblanc G. The electrochemical proton gradient in Mycoplasma cells. Eur J Biochem.; 113(3):491-498 (1981).
69. Le Grimellec C, Lajeunesse D, Rigaud JL. Effects of energization on membrane organization in mycoplasma. Biochim Biophys Acta.;687(2):281-290 (1982).
70. Yoch DC, Carithers RP. Bacterial iron-sulfur proteins. Microbiol Rev. ;43(3):384-421 (1979).
71. Hall DO, Rao KK, Cammack R. The iron-sulphur proteins: structure, function and evolution of a ubiquitous group of proteins. Sci Prog. 62(246):285-317 (1975).
72. Reizer, J., I.T. Paulsen, A. Reizer, F. Titgemeyer, and M.H. Saier, Jr. Novel phosphotransferase system genes revealed by bacterial genome analysis: the complete complement of pts genes in Mycoplasma genitalium. Microbial Comp. Genomics 1: 151-164 (1996).
73. Reizer, J., V. Michotey, A. Reizer, and M.H. Saier, Jr. Novel phototransferase system genes revealed by bacterial genome analysis: unique, putative fructose- and glucoside-specific systems. Prot. Sci. 3: 440-450(1994).
74. Chen, Y., J. Reizer, M.H. Saier, Jr., W.J. Fairbrother, and P.E. Wright. Mapping of the binding interfaces of the proteins of the bacterial phosphotransferase system, HPr and IIAglc. Biochemistry 32: 32-37 (1993).
75. Reizer, J., T.M. Ramseier, A. Reizer and M.H. Saier, Jr. Novel phosphotransferase genes revealed by bacterial genome analysis: A gene cluster encoding a phosphotransferase system permease and metabolic enzymes concerned with N-acetylgalactosamine metabolism. Microbiol. 142: 231-250 (1996).
76. Hvorup, R.N., A.B. Chang, and M.H. Saier, Jr. Bioinformatic analyses of homologues of the bacterial L-ascorbate PTS permeases. J. Mol. Microbiol. Biotechnol (2004).
77. Greenberg, D.B., J. Stülke, and M.H. Saier, Jr. Domain analysis of transcriptional regulators bearing PTS-regulatory domains. Res. Microbiol. 153: 519-526 (2002).
78. Hvorup, R.N., B. Winnen, A. Chang, Y. Jiang, X.-F. Zhou, and M.H. Saier, Jr. (2002). The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) flippase superfamily. European J. Biochem. 148: 3760-3762.
79. Axelsen, K.B. and M.G. Palmgren. (1998). Evolution of substrate specificities in the P-type ATPase superfamily. J. Mol. Evol. 46: 84-101.