Evidence of Contemporary Modern Human Evolution Contained Within the Human Genome
Platter, Branden E.
Abstract
The question of whether or not modern humans are still evolving is being asked more than ever with the vast amount of technology, culture and medicine which have the ability to buffer environmental stressors that would otherwise drive evolutionary adaptations in humans. The purpose of this paper was to determine, in spite of these "advances," if modern humans are still evolving by searching for proof within the human genome. The three prime examples of current evolution discussed are: certain populations' resistance to malaria, lactase persistence and resistance to HIV, each of which are adaptations regulated by mutant alleles. Each adaptation was broken down by the different mutations responsible for that adaptation. In order to determine if these mutations are, in fact, evidence of modern evolution, they were each subjected to a strict list of criteria necessary to be deemed adaptive and evolutionary. In all cases except one, the mutations passed the criteria and are considered proof that modern humans are still evolving. It is concluded that even though modern humans have the ability to buffer environmental stressors that would typically cause evolutionary changes, not all of these can be buffered and as a result, the human genome is evolving to adapt to these stressors.
Citation
Evidence of Contemporary Modern Human Evolution Contained Within the Human Genome. Platter, Branden E. . Lethbridge Undergraduate Research Journal. Volume 4 Number 1. 2009.
Introduction
An increasingly common opinion is that humans have hit an evolutionary peak or adaptive plateau. A combination of complex culture, technology, and biomedical advances has effectively buffered many human populations against many of the selective forces that would otherwise drive continual evolution of our species. “Most laypeople tend to assume that humans are the pinnacle of evolution and that we have stopped evolving (Balter, 2005a).” This statement by Huntington Willard, director of the Institute for Genome Sciences and Policy at Duke University, is indicative of one of the major questions in the field of human biological variation and human evolution. Are humans still evolving?
The purpose here is to explore multiple examples of evolution within the human genome, primarily focusing on three specific examples of selective sweeps that are in progress at the present time: resistance to malaria, lactase persistence, and resistance to HIV. By exploring these topics and the ability of humans to adapt to these conditions, it will show that not only has human evolution continued to the present day, but will continue indefinitely.
Defining Evolution
In its most basic definition, evolution is described as a change in the frequency of alleles in a given population over time (Freedman & Herron, 2004). Evolutionary theory tells us that the majority of evolution is brought about by selective pressures acting upon a given population. Selective pressures are responsible for the evolution from australopithecines to early Homo species to modern humans. Yet, there is a current question about the effects of modern humans' culture on these selective pressures and whether our culture is counteracting these selective pressures to a degree that it has prevented the continual evolution of our species.
As a major evolutionary force, positive natural selection works to promote alleles coding for traits that give an individual more reproductive fitness and negative natural selection works to eliminate alleles coding for traits that are harmful to an individual (Nielsen et al., 2007), so the expectation is to see selection working on the genes that cause these respective traits. All things being equal, over time, we should see the extinction of alleles coding for disadvantageous traits and fixation of those coding for advantageous traits. It is in the genome that we can discover the answer to the question of whether or not modern humans are still evolving.
Genetic mutations are the sole creator of new genetic material and the cause of variability in the human genome. Single nucleotide polymorphisms (SNPs) are so common (reflecting the variability of the human genome) that they occur at a frequency of 1 in 1,000 base pairs, however, about 75% of these SNPs are in non-coding portions of the DNA or are silent mutations (do not change which amino acids are coded for) (Shastry, 2007). The importance of these mutations is the allowance of variability for human adaptability. The ability of an organism's genome to create mutations within its sequence is self-preservation of genetic variation leading to adaptability. While medical technology may have the ability to buffer against the effects of adverse alleles and material culture may have the ability to buffer against selective pressures, mutations will provide a continual mechanism of genetic changes, continually opening doors to possible frequency increases, thus evolution of the human genome.
To determine whether or not humans are still evolving, we must look at the genome and determine if there are documented cases of changes in allele frequencies. A change in frequency of alleles would signify that the allele is being favored (shown by an increase in frequency) or that it is being selected against (shown by a decrease in frequency). The methods for estimating whether or not a gene is being selected for often varies depending on the population and gene being studied, but they all follow one basic premise: If positive selection acts on protein-coding genes and selection is recurrent, selection can be detected by an increased rate of mutated allele frequencies (Nielsen, 2007). Selective sweeps, where a new mutation becomes fixed so quickly that physically linked alleles also become fixed or lost with it, eliminating variation at tightly linked loci and creating an excess of alleles of very low and very high frequency at more distant loci (Nielsen, 2007; Williamson et al., 2007), are the primary focus of these allele frequency tests. These tests look at SNPs analyzing the frequency of alleles connected to the “favored allele” based on distance from the locus.
Other tests look for evidence that allele mutations responsible for changing the protein the allele codes for have been favored over those that cause no change (Balter, 2005b). Some tests look at gene frequencies within a population or between populations and examine whether they are more common than would be expected by genetic drift. A more recent approach compares an allele frequency in a population with the genetic diversity within the haplotype to which it belongs (Balter, 2005b). Knowledge of the human genome allows us to estimate both point mutation rates and deleterious mutation rates per generation. According to Reed and Aquadro (2006), multiple studies have given conservative estimates of up to four deleterious mutations per generation, but more recent studies have raised that estimate to about ten.
Current estimates by Williamson et al. (2007) show that there are approximately 101 regions of the genome that show evidence of a recent selective sweep (Appendix I). These regions contain both the allele being selected for and the alleles within close proximity that increase as a result of selection on the original allele. For this study, populations were divided into categories of African-American, European-American, Chinese, and combined. The African-American sample showed only nine regions of recent selective sweeps (Williamson et al., 2007). This coincides with what we should expect to see. With modern humans originating in Sub-Saharan Africa, those populations have had at least 100,000 years of adaptation to the selective pressures of that environment. The European-American and Chinese populations (with 26 and 57 regions respectively showing recent selective sweeps) have a history of migration out of the Sub-Saharan environment into new, varied environments. We would expect to see more current adaptation in these areas given human populations' more recent move into these environments providing a new set of selective pressures to adapt to. There is also a trend of increased selective sweeps in subpopulations which may give some insight into the importance of local micro-environments in human adaptation (Williamson et al., 2007).
“[S]cientists point out that in developed countries, culture, technology, and especially medical advances have changed the evolutionary rules, from survival of the fittest to the survival of nearly everyone… [y]et millions of people in developing countries continue to live under the combined stresses of poverty and disease. Under these conditions, even skeptics of ongoing human evolution agree that natural selection may be favoring genes that confer resistance to disease or enhance reproductive fitness in other ways (Balter, 2005b: 234).” Multiple specific examples of selective sweeps are currently in progress throughout different populations. “There is evidence that the population prevalence of some human phenotypes, such as resistance to malaria or lactose tolerance in adulthood, results from natural selection in response to idiosyncratic conditions (Barreiro et al., 2004: 340).”
Criteria for Determining Evolution
In the search for proof of continual human evolution in the genome, we must first decide what will constitute proof of evolution. Since evolution is a change in the frequency of alleles in a population over time, and we expect to see this as a result of adaptation to environmental stressors, the criteria should pertain to the frequency of the mutative alleles and the relationship of these mutations to the region's selective pressures. The criteria for determining adaptive evolution of an allele are:
- The mutative allele confers an identifiable benefit to the individuals who carry it,
- In the region(s) that the mutative allele is present, the benefit of possessing the trait outweighs the cost of possessing it,
- The mutative allele shows an increase in frequency from a previous time,
- Multiple mutative alleles confer the same benefit to the same stressor,
- The stressor that is being combated by the mutative allele cannot be fully buffered by cultural or behavioral methods, and
- The frequency of the mutative allele is greater in areas where the stressor is present than in areas where the stressor is not present or the allele confers no benefit.
While it is not necessary for each mutation to adhere to every criterion, a minimum of four of the six criteria are required in order for the given mutation to be considered convincing evidence for continual evolution.
Current Evidence
Examining modern genetic mutations within the human genome can provide evidence for the current evolution of modern humans. Many mutations provide a beneficial adaptation to a selective force so by identifying these selective forces we can determine mutations that confer an adaptation to them. Some of the most prominent contemporary selective forces are high altitude hypoxia, brain size, malaria, lactose intolerance, HIV, and western biomedicine. Mutations that are currently at a high enough frequency to be considered a polymorphism are subjected to the previous criteria to determine whether or not they are consistent with requirements for determining evolution.
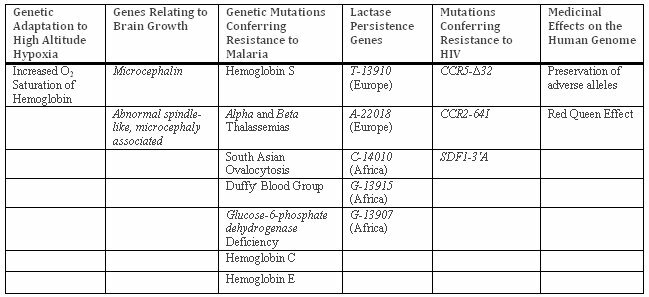
Table 1: Evidence of Evolution within the Human Genome
High Altitude Hypoxia
With the migration of human populations into high altitudes, a new set of selective pressures are at work in populations of the Andean and Tibetan highlands. The decreased barometric pressure with increased altitude leads to a lowered amount of oxygen intake per breath. This means a decreased amount of oxygen can be disbursed to body tissues where it is needed for mitochondrial metabolism (Beall, 2001). This decrease of oxygen in the blood, called hypoxia, impairs physical and mental performance. It can cause diseases as mild as acute mountain sickness (headache, lightheadedness, fatigue, and insomnia) to diseases as severe as high-altitude pulmonary or cerebral edema (tachycardia, papilledema, coma, or death) (West, 2004). The affects of hypoxia can be reduced by acclimatization (hyperventilation, polycythemia, and pulmonary vasoconstriction), but cannot be abolished completely (West, 2004; Beall, 2001).
Increased Oxygen Saturation of HemoglobinRecent studies have begun assessing the possible genetic adaptations that these populations have acquired or are currently acquiring to adjust to this low-oxygen environment. One of these studies by Beall (2004) has focused on the fertility of the Tibetan highland populations and the possibility of an allele coding for increased oxygen saturation in hemoglobin. High altitude environments, with their low-oxygen availability, create susceptibility of individuals to hypoxia. Currently, the locus containing the gene for oxygen saturation is unknown, but a series of data analysis methods has allowed Beall's research team to estimate fertility based on probable genotypes.
While the number of conceptions and live births did not vary among the genotypes, the mortality rate of the offspring did. The number of infants that died varied from a mean of 0.48 for the high oxygen saturation genotype to a mean of 2.53 for the low oxygen saturation genotype. The number of children living (which is important due to evolution's dependence on individuals reaching a reproductive age) varied from 3.79 in the high oxygen saturation genotype to 1.64 in the low oxygen saturation genotype (Beall, 2004). We can infer that a selective sweep is in progress in the Tibetan highlands given the higher fitness of offspring from high oxygen saturation genotypes.
Brain Growth
We often view natural selection as a process acting primarily on physical aspects of an organism and forget that selection can also select for cognitive abilities. For this reason, it seems pertinent to focus first on one of the key aspects that have been used to separate hominid species: brain size. Many factors have been suggested as selective forces behind the evolution of cognitive abilities. Reed and Aquadro (2006) cite social competition as one of the primary selective forces behind the evolution of human cognitive abilities; others have said that it is the necessity to remember locations of important resources in times of scarcity that has led to an increased brain size. Dunbar, a prominent researcher in this area, has described a “Machiavellian Intelligence” explanation for increased brain size. He cites both group size and social complexity, including both quantitative and qualitative aspects of social groups, as the driving force behind all primate neocortex growth (Dunbar & Shultz, 2007). Regardless of the mechanisms responsible (and there are many proposed), there is a definite increase in brain size through hominid evolution.
Microcephalin and Abnormal spindle-like, microcephaly associated (ASPM) Regulating Brain GrowthAs the primary mechanism associated with cognitive abilities, we would expect to see selection in the human brain, but common thought has been that the human brain has reached an evolutionary peak. Recent studies (Evans et al., 2005; Mekal-Bobrov et al., 2005) focusing on two genes thought to regulate brain growth suggest that the human brain may still be evolving. Six loci contain genes for which recessive mutations lead to microcephaly (MCPH), named MCPH1- MCPH6. The genes microcephalin (MCPH1) and abnormal spindle-like, microcephaly associated (ASPM, MCPH5) are two of these genes associated with microcephaly (Dobson-Stone et al., 2007). The frequency of the alleles MCPH1 G37995C and ASPM A44871G have come under recent studies.
Evans et al. (2005) and Mekal-Bobrov et al. (2005) studied a collection of human cells representative of global human diversity and found that, for each gene, there was an allele with surprisingly high frequency. These frequencies were high enough to be considered above the realm of genetic drift or gene flow suggesting that this frequency is the result of natural selection. By estimating mutation rates, Evans et al. (2005) and Mekal-Bobrov et al. (2005) were able to estimate time frames for when each allele arose. MCPH1 seemed to arise around 37,000± 23,000 years ago (around the time of the explosion of symbolic behavior in Europe) and the ASPM allele is thought to have arisen 5,800± 5,500 years ago (around the time cities arose in the Near East). Whether it can be concluded that the rise of these genes directly contributed to the rise of symbolic behavior or development of the first cities is purely conjecture.
A study done by Dobson-Stone et al. (2007) showed conflicting evidence. This study showed that neither MCPH1 G37995C nor ASPM A44871G had a significant effect on brain size or the relative size of the cerebral cortex. However, more efficient techniques may provide a better insight into the relationship of these genes to brain size. More research is needed on these genes and their possible connection to cognitive ability and the development of culture.
Malaria
The invention of agriculture caused a dramatic increase in the clearing of forests for farmable land (Balter, 2005b). The increase in the area of flat, root-free land created the optimal situation for standing water to accumulate providing an increase in breeding areas for the malaria-carrying Anopheles gambiae species of mosquito (Wiesenfeld, 1967). The invention of agriculture also decreased the mobility of human populations tying individuals to their lands. Humans were also forced to share their land with many animals, both domesticated and wild (Cockburn, 1971). As the optimal environment for the Anopheles mosquitoes to breed increased and humans became adopted a sedentary lifestyle in close proximity to animals, the world saw an endemic explosion of malaria, caused by the cultural “advance” of civilization (Balter, 2005b).
Malaria is most often caused by the parasites Plasmodium falciparum and Plasmodium vivax. After the initial injection of the parasite into the subcutaneous tissue, the parasites move to the liver's hepatocytes (protein synthesizing cells) where they develop into tens of thousands of merozoites (asexually produced daughter cells of parasites). These merozoites then infect red blood cells (RBCs), by inducing a vacuole derived from the RBCs plasma membrane, where they continue to multiply. This is a quick process taking only 48 hours and produces 20 merozoites per mature parasite (Miller et al., 2002). Tishkoff et al. (2007) describes the RBC as the hub of the parasitic malarial life cycle providing food, shelter, and access for the infected red blood cells to reach non-infected RBCs. By the process of breaking down hemoglobin to create room to grow, the parasites release toxic by-products, particularly iron (Kwiatkowski, 2005). The disease is characterized by symptoms including severe anemia (compromising oxygen delivery to body tissues), fever, nausea, flu-like symptoms, organ failure, dehydration, hypovolemia (decreased volume of blood plasma), coma and even death (Greenwood et al., 2005; Miller et al., 2002). P. falciparum also causes infected RBCs to adhere to blood vessel linings causing blood clots (Miller et al., 2002). The basic process of malarial infection follows the basic scheme of most major infectious diseases characterized by Miller et al. (2002: 674): “rapid expansion of infected RBC mass, destruction of both infected and uninfected RBCs, microvascular obstruction, and inflammatory processes that combine to lead to reduced tissue perfusion.”
While both P. falciparum and P. vivax cause severe anemia, only P. falciparum causes cerebral malaria, hypoglycemia, metabolic acidosis, respiratory distress, and adhering of infected RBCs to blood vessel linings causing blood clots (Greenwood et al., 2005; Miller et al., 2002). P. falciparum can invade RBCs by numerous pathways making most malarial populations susceptible to this form. P. vivax is limited to invasion of Duffy + blood group RBCs and as a result, has nearly disappeared from West Africa where the Duffy + blood group is not present (Miller et al., 2002).
According to Hay et al. (2004), the global distribution of malaria before mosquito control efforts began in the early 1900s spanned more than 50% of the global land surface area and was present on all continents except Antarctica. A century of regulation efforts to eliminate mosquito breeding sites in developed areas has decreased that percentage to about 27% and eliminated it from most of the developed regions of the world. However, P. falciparum still causes an estimated half a billion episodes of disease every year (Kwiatkowski, 2005). Despite control efforts, contemporary problems continue to promote the infection and transmission of malaria. Civil disturbances cause the collapse of treatment programs and crowding of refugees in unquarantined quarters. An increased frequency of travel subjects individuals of non-malarial regions to infection. Climatic factors such as warming, drought and floods can increase the chance of transmission of malaria (Greenwood et al., 2005). It is expected that this endemic of malaria would create a selective force on human populations in areas where control has not been possible. This is what we are seeing with populations that reside in malarial environments (Appendix II).
Because the RBC is the hub of malarial infection, the expectation is that selection would affect genes coding for RBC structure or function. Over the last 10,000 years, malaria has resulted in hundreds, if not thousands, of genetic variations that exhibit some sort of resistance to the disease (Williams, 2006b). Several contemporary alleles provide resistance to malaria including sickle-cell hemoglobin, α- and β-thalassemias, South Asian ovalocytosis, Duffy negative blood group, the alleles A- and Med of the glucose-6-phosphate dehydrogenase gene (G6PD), Hemoglobin C, and Hemoglobin E (Balter, 2005b; Williams 2006a,b). Their frequencies in correlation to regions of endemic malaria provide evidence that they are examples of adaptive evolution of the human genome.
Hemoglobin SThe best understood of the hemoglobinopathies is sickle-cell hemoglobin or hemoglobin S (HbS). This point mutation in the beta hemoglobin chain causes the hemoglobin, HbS, to polymerize at a low oxygen concentration giving RBCs a deformed sickle shape (Kwiatkowski, 2005). It is still unclear exactly how sickle hemoglobin prevents infection by malaria but heterozygotes for this allele (HbAS, where HbA is normal hemoglobin allele) are protected from severe and mild malaria (Roberts & Williams, 2003). HbAS individuals experience a >90% resistance to severe and lethal malaria and a 50% resistance to mild clinical attacks (Williams, 2006a, b). While individuals homozygous for the ancestral HbA allele are susceptible to malaria, individuals homozygous for the HbS allele experience sickle-cell anemia, therefore heterozygocity of the sickle-cell trait is an example of heterosis, where individuals with the heterozygous genotype exhibit a greater fitness than individuals with either homozygous genotype (Tchuenche, 2005).
Alpha and Beta ThalassemiasA set of diseases affecting the synthesis of hemoglobin, known as thalassemias, have also shown resistance to malaria. These syndromes are known as alpha (α)- and beta (β)-thalassemias for their underproduction of their respective α- and β-globins (Roberts & Williams, 2003; Williams, 2006a). Both groups have been observed in high frequencies in regions of malaria. The first group, α-thalassemia, is only beneficial in a heterozygous form. Homozygosity for the allele is often fatal. Occasionally, only one of the two genes coding for α-globin are disrupted and some α-globin is produced. This is referred to as α+ thalassemia and homozygotes experience only mild anemia. This disease increases the RBC's susceptibility to invasion by malaria. Children with α+ thalassemia are actually more susceptible to contracting malaria than non-thalassemia individuals (Kwiatkowski, 2005). It is thought that by predisposing individuals to malaria early in life, they can develop a higher immunity to the disease (Roberts & Williams, 2003). Unlike HbAS, α-thalassemia seems to only affect severe malaria anemia (Williams, 2006a).
South Asian OvalocytosisSouth Asian ovalocytosis (SAO) is caused by heterozygosity of a 27-basepair deletion in the gene encoding the RBC membrane protein band 3 (SLC4A1Δ27) (Williams, 2006b). The result of this is an oval shape of the RBCs (Kimura et al., 2006). While homozygosity for the SLC4A1Δ27 deletion is believed to be fatal, the high frequency of heterozygosity in malaria endemic regions reflects its beneficial function as a malaria resistant (Williams, 2006b). Not all cases of ovalocytosis are caused by the B3Δ27 deletion, but both those caused by it and those not caused by it seem to have some degree of protective effects against malaria (Kimura et al., 2006). More research is still needed on this topic.
Duffy Blood GroupThe Duffy blood group characterized by the chemokine receptor FY, encoded by the FY gene, has been associated with resistance to malaria (Kwiatkowski, 2005). The product of this gene is a glycoprotein which spans the cells membrane and has an additional extra cellular and intra cellular terminal domain. The Duffy antigen serves as the receptor for P. vivax so negative homozygosity for this allele confers resistance to the parasite (Langhi Jr. & Bordin, 2006).
Glucose-6-phosphate dehydrogenase DeficiencyThere also seems to be a correlation between glucose-6-phosphate dehydrogenase (G6PD) deficiency in RBCs and protection against malaria. The mutant A- and Mediterranean (Med) alleles cause a deficiency of G6PD in RBCs. It is thought that the protection against malaria involves early phagocytosis of G6PD-deficient parasite-infected RBCs at twice the efficiency of normal RBCs and reduced parasite replication in erythrocytes (Kwiatkowski, 2005; Williams, 2006a). This efficient phagocytosis of infected RBCs is a result of the impaired anti-oxidant defense in G6PD deficient RBCs (Williams, 2006b). However, as an adaptation to G6PD deficiency, the parasites have developed the ability to produce their own G6PD (Kwiatkowski, 2005).
The distributions of the two G6PD alleles are highly correlated with areas of endemic malaria. In tropical Africa, almost 90% of G6PD deficiency is a result of the A- allele which is also frequent in North and South America, the West Indies, and regions where people of African origin are present. The Med allele is common in all countries surrounding the Mediterranean Sea (Cappellini & Fiorelli, 2008).
Hemoglobin C/Hemoglobin EThough the research at this point is fairly limited, it seems that other hemoglobinopathies show evidence of protection against malaria. Hemoglobins C and E have exhibited resistance to the parasite. Heterozygotes for HbE (HbAE), common in Southeast Asia, has shown a high resistance to invasion by the parasites while homozygotes (HbEE) exhibit a lower resistance than the heterozygote. In heterozygotes, only 25% of the RBCs are susceptible to invasion by the malaria parasites (Chotivanich et al., 2002). HbC has also proven effective as a protector. In clinical tests heterozygotes (HbAC) showed a 29% reduced risk of malaria while homozygotes (HbCC) showed a 93% reduction in malaria (Fairhurst et al., 2003; Roberts & Williams, 2003; Williams, 2006a). In clinical tests by Fairhurst et al. (2003) HbCC and HbAA RBCs showed equal levels of invasion by malaria parasites. HbCC RBCs exhibited a substantial degree of parasite death within the cell. The parasite death rate within the cell, at about 50%, limits the multiplication of the malaria parasites, controlling the severity of the disease.
Hemoglobin C and hemoglobin E are both less deleterious than hemoglobin S. This is due to the early destruction of HbS RBCs and the clogging of these sickled cells in the microcapillaries (Kate & Lingojwar, 2002). More research is needed on both HbC and HbE and their correlation to malaria resistance.
Lactose Intolerance
Lactose (β-D-galactosyl-D-glucose) is a disaccharide that is found only in mammalian milk. While humans posses the largest quantity of lactose among all mammals at 7g per 100mL, cows' milk has nearly half that with 4.6g per 100mL (Schaafsma, 2008). In areas of drought and arid climate, milk can be an important source of nutrients and water (Gibson, 2007). Lactase-phlorizin hydrolase (LPH) is a β-galactosidase enzyme found in the small intestine where it hydrolyzes lactose from milk into its monosaccharide components, glucose and galactose, sugars that are easily absorbed into the bloodstream (Tishkoff et al., 2007; Schaafsma, 2008). Most humans stop producing lactase after weaning, but a growing population of individuals have continued to produce lactase into adulthood (Balter, 2005b; Shastry, 2007; Tishkoff et al., 2007). Lactase nonpersistence is the ancestral state and populations of high lactase persistence frequency correlate to the invention of agriculture, when milk from domesticated animals became available to drink by humans (Hollox, 2005).
The nutritional level of lactose provides a selective advantage to those who retain the ability to digest it. While high levels of blood glucose increase the risk of cardiovascular disease, lactose has a relatively small glycemic index (Schaafsma, 2008). This means that since lactose is usually not fully digested and the galactose monosaccharide needs additional conversion into glucose in the liver, lactose can decrease an individual's blood glucose level (Schaafsma, 2008). Undigested lactose also acts like a dietary fiber increasing the water content of stools and reducing transit time in constipated subjects. Undigested carbohydrates from lactose that are fermented in the colon enhance the absorption of the minerals calcium and magnesium (Schaafsma, 2008).
Individuals who do not retain the LPH enzyme can still consume lactose but only at a quantity of 11g per day (equaling one or two portions) when this amount is spread throughout the day and ingested with food. Any more than 11g will often cause bloating, cramps, gas formation and diarrhea as a result of bacterial fermentation of the undigested lactose in the colon (Schaafsma, 2008). It is highly beneficial for individuals to be able to process lactose for its nutritional value given that not only is the ability to digest lactose adaptive in regions of nutritional stress, the inability to digest it can be harmful, increasing diarrhea and dehydration.
Individuals that retain the enzyme LPH are said to have the ‘lactase persistence trait.’ It is estimated that about 20% of the global population maintain lactase persistence throughout their lifetime (Schaafsma, 2008). The distribution of these groups of individuals is largely correlated with the spread of domesticated cattle out of the Near East (Balter, 2005b; Tishkoff et al., 2007). As would be expected with a correlation to the spread of domesticated cattle, the more dependant on pastoralism a population is, the higher the frequency of lactase persistence in that population. This is highest in northern Europe populations at >90% lactase persistence, decreasing to ~50% across southern Europe and the Middle East, ~5-20% in West African populations and a mere ~1% in Chinese populations (Tishkoff et al., 2007). Since dairying is thought to have originated around 10,000 years ago, the rapid increase in lactase persistence gene in only 400 generations (Hollox, 2005) is outside the realm of genetic drift.
Genetic Mutations Conferring Lactase PersistenceLactase persistence gene (LCT) is a dominant Mendelian trait located at 2q21 locus. Tishkoff et al. (2007) showed that two SNPs are associated with lactase persistence in Europeans: T-13910 and A-22018 alleles. A cross-study of lactase persistence in African regions showed SNPs C-14010, G-13915, and G-13907 to be associated with lactase persistence, not either of the two T-13910 or A-22018 alleles. The two polymorphisms associated with LCT are located 100 base pairs apart in intron 13 of the MCM6 gene located 14 kilobases away from LCT (Gibson, 2007). This difference in SNPs responsible for lactase persistence in different geographical regions suggests that lactase persistence has been selected for independently in different regions.
The fact that this trait has been selected for independently in different regions is a good indication that it is the result of selective pressure on humans. This has been called the best example of convergent evolution in humans (Check, 2006). Were the lactase allele negative, it would be expected to be short-lived and eventually face extinction as selective pressures would work against it. Were it neutral, we would not expect to see an increase in allele frequency, rather a plateau. What is seen with lactase persistence is a move toward fixation as, at least certain regions such as Europe and selected populations in Africa (Tishkoff et al., 2007), have acquired extremely high frequencies of the allele. The ability to process lactose-rich milk and harness its nutritional value may have allowed people to survive times of drought and resource scarcity since those with lactase persistence could drink milk without the risk of diarrhea and increased dehydration (Check, 2006). Lactase persistence has been estimated to give a fitness advantage of 5-10%, one of the strongest known selection differentials in human variation (Gibson, 2007).
HIV/AIDS
One of the most well known contemporary epidemics plaguing modern human populations is HIV/AIDS. What is currently thought of as an incurable disease pertinent to all populations may be in the early stages of a selective sweep. Nearly 40 million humans carry this virus which has a fatality rate of close to 100% (Rambaut, 2004).
Human immunodeficiency virus's (HIV) similarity to simian immunodeficiency viruses (SIVs) is regarded as evidence of human contraction from nonhuman primates. Both HIV viruses (HIV-1 and HIV-2) are more closely related to different SIVs suggesting different origins in the human population. The more common HIV-1 resembles SIVcpz, found in Pan troglodytes troglodytes and Pan troglodytes schweinfurthii of Western and Central Africa while HIV-2 more closely resembles SIVsm found in Cercocebus atys of West Africa (Rambaut, 2004). The three different subgroups of HIV-1 (M, N, and O) are thought to represent different origins of HIV-1 to humans from chimpanzees (Rambaut, 2004).
Controversial estimates have suggested an HIV-1 M group origin as recent as the 1930s ± 10 years (Rambaut, 2004). While this may seem like sufficient time to establish cultural and behavioral defenses to the virus (vaccines, antibiotics, etc.), certain characteristics of the HIV viruses prevent these methods from being effective. HIV is one of the fastest evolving organisms due to its high mutation rate, high replication rate (viral generation time of ~2.5 days with ~10 10 - 10 12 new virions (mature virus particle) each day), frequent recombination (three recombinations per genome per replication cycle) and natural selection (Rambaut, 2004). The virus is also capable of rapidly fixing mutations that confer evasion of immune responses, occurring at a rate of one adaptive fixation every ~2.5 months (Rambaut, 2004).
To attach to CD4 + T lymphocyte cells (a key component of the human immune system), the HIV virus utilizes chemokine co-receptors CCR5 and CXCR4, initially using the CCR5 receptor in early years, but broadening to use both CCR5 and CXCR4 in later years of infection (Rambaut, 2004). Highly active antiretroviral therapy (HAART), which is a combination of drugs acting against different aspects of the virus' life cycle, had been thought of as a cure for HIV, though more recently it has proven ineffective (Rambaut, 2004). Since behavior and culture have been unable to buffer the HIV virus, mutations in these co-receptors have provided a genetic mechanism of combating the infection at a global level (Appendix III).
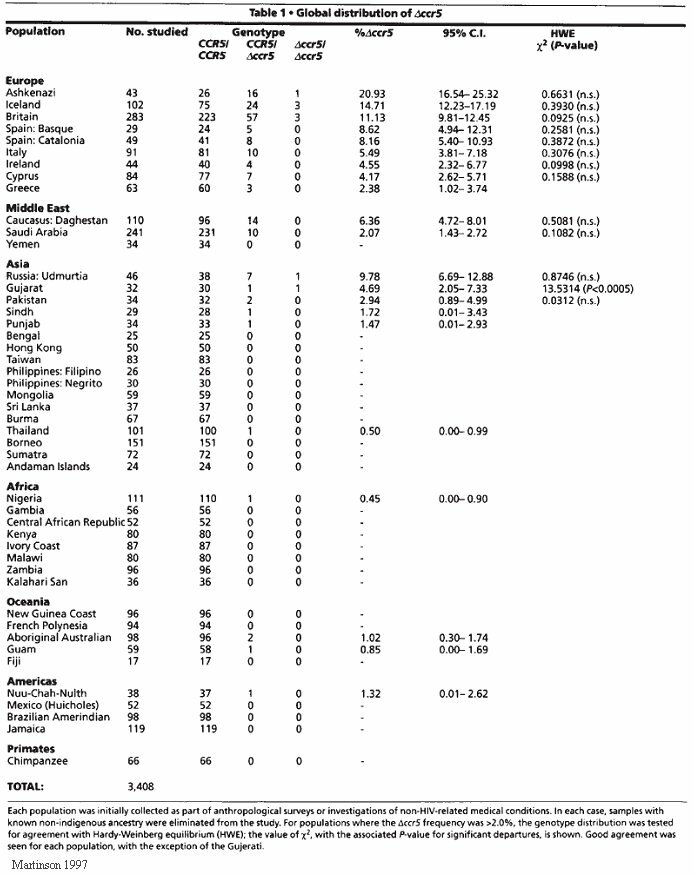
CCR5-Δ32In the mid 1990s, it was discovered that a deleterious mutation in the chemokine receptor 5 (CCR5) gene provides protection against HIV. The recessive mutation, CCR5-Δ32, deletes the CCR5 receptor from CD4 + T lymphocyte cells providing protection from this attachment in individuals who are homozygous for the allele. Individuals who are heterozygous (CCR5-Δ32/+) for the allele may confer partial resistance though it is more likely that heterozygosity postpones the progression of HIV to AIDS by an additional 2-3 years (Martinson, 1997; Galvani & Novembre, 2005; Smith et al., 1997). Researchers originally dated the CCR5-Δ32 mutation to about 700 years ago (Balter, 2005b) which suggests that it was a different selective pressure, either the plague or smallpox, on the gene that favored the mutation. Newer estimates, based off the presence of the CCR5-Δ32 mutation in Bronze Age skeletons (2,900 BP), implies a different selective agent for the mutation (Hedrick & Verrelli, 2006), though one has yet to be proposed. Based on its European frequency, the CCR5-Δ32 deletion would be estimated at 127,500 years old if it had not undergone natural selection (Galvani & Novembre, 2005). This highly suggests the presence of a selective force occurring at some time since the original mutation. According to Martinson et al. (1997), this polymorphism frequency in Europe is consistent with what would be expected from genetic drift acting on a neutral polymorphism and that the introduction of HIV-1 into Europe has been too recent to have an effect on the distribution of CCR5-Δ32. However, this does not negate the beneficial properties of the mutation in resistance to HIV.
Usually, loss of gene function, as with the CCR5-Δ32 deletion, falls under negative selection, eliminating it from the genome. Given that HIV uses CCR5 as a means to enter T lymphocyte cells, it seems that the loss of gene function is advantageous in this instance (Hedrick & Verrelli, 2006). This case illustrates the ability for even deleterious mutations to be favored in the genome. This mutation has an allele frequency average of 10% of European populations (translating to a homozygote frequency of 1%), 2-5% throughout the Middle East and Indian subcontinent, but is extremely rare throughout the rest of the world (Appendix IV) (Martinson, 1997; Galvani & Novembre, 2005). The isolated cases are most likely a result of gene flow from Europe. However, this subject should be monitored as gene flow introduces the allele to isolated areas and leads to increased frequency and selective sweeps around the world.
CCR2-64IA nucleotide substitution on the CCR2 gene (CCR2-64I) was originally thought to confer resistance to HIV. While CCR5-Δ32 is found almost exclusively in European populations, CCR2-64I has been found in every ethnic group tested (Smith et al., 1997). A study by Smith et al. (1997) showed an allele frequency of 0.098 in Caucasians, 0.151 in African Americans, 0.172 in Hispanics, and 0.250 in Asians. There were no differences in allele frequency between HIV infected and exposed, uninfected individuals. These frequencies conform to Hardy-Weinberg expectations, eliminating a correlation between allele carriers and HIV infection rates (Smith et al., 1997). Different allele frequencies have been found between HIV infected individuals with a slow progression to AIDS and those with a quick progression to AIDS. The CCR2-64I allele frequency showed a 30 to 80% increase among groups with slow progression or no progression to AIDS at all. Heterozygous (CCR2-64I/+) individuals also showed a slowed progression to AIDS (Smith et al., 1997).
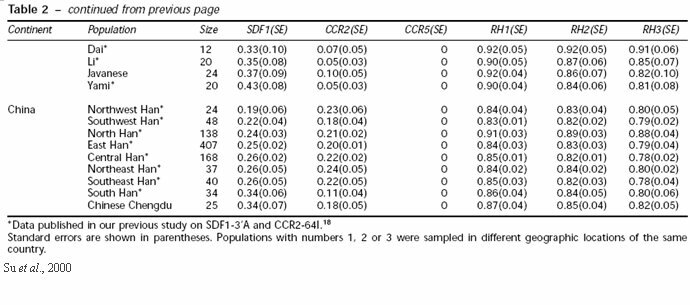
SDF1-3'ARecent research has explored the effects of a polymorphism in the 3' untranslated region of the stromal cell-derived factor 1 (SDF1/CXCL12) gene on either resistance to HIV-1 or delayed progression to AIDS (Reiche et al., 2006). The mutant recessive allele named SDF1-3'A is the only natural ligand of the co-receptor CXCR4. Individuals who are homozygous for this allele have shown resistance to AIDS. The belief is that the allele restricts the emergence of X4 tropic HIV-1 strains by overproducing SDF1/CXCL12 which then binds to and blocks the CXCR4 receptor from attachment by the X4 virus (Reiche et al., 2006).
Current frequencies of the SDF1-3'A allele are much higher than the CCR5-Δ32 allele being exceptionally high in Oceania and reaching as high as 72% in New Guinea highlanders. Other populations range from 3% to 43%, however the allele is almost absent in African and Southeast Asian populations (Su et al., 2000). While individuals who are homozygous for the SDF1-3'A allele showed an increased resistance to HIV-1, other studies have come up with conflicting data so more research is needed on the effects of the mutant genotype on the resistance to HIV-1.
Biomedical Effects on the Human Genome
It is important to note that evolution is often used synonymously with adaptation, but this implies that evolution is always positively adaptive. This is not the case. The persistence or possible increased frequency of disease susceptible SNPs is in and of itself evolution. It is a change in the frequency of alleles within a population. Evolution is not always adaptive, but whether adaptive or not, the human genome is always evolving.
One of the primary contributors to the supposed halting of continual evolution is the degree to which western biomedicine has influenced humans' ability to buffer the effects of harmful genes. The evolution of biological and medical understanding has allowed the increased fitness of those individuals who without the help of medicine would not have reached reproductive age. While at a cultural level this is seen as a great accomplishment, the effects on the human genome seem to be of little consequence. Many of these conditions are the result of adverse alleles which selection is trying to work against. By providing pre- and postnatal care to these individuals, we are allowing the preservation of deleterious alleles; and if the individual reaches reproductive age and reproduces, biomedicine has directly caused the increase in frequency or maintenance of frequencies of adverse alleles in the population. Without medical technology, these individuals would not reach a reproductive age and in the absence of any other fitness benefits from the allele, selective pressures would eliminate those adverse alleles from the genome.
A study on natural selection in an Australian population gives us an example of how medical intervention can reduce the effects of selection. According to Stephan and Henneberg (2001: 635):
“This medical intervention [modern medicine] frequently allows individuals to survive and reproduce. Hence, it is possible for people who are genetically and phenotypically less adapted to the environment to pass their genes on to the next generation. This may increase genetic load, since unfavorable genotypes can accumulate. Although medical treatment has increased the average time a human can expect to live, the associated accumulation of unfavorable genes in the human population has probably decreased the human capacity to survive without medical intervention. Many humans have, therefore, come to rely on medicine for their health. This dependency may increase in the future as more unfavorable genes accumulate in the gene pool.”
Medical treatment exerts selective pressures on pathogens against which the human immune system is fighting. One of the most common examples of this is Streptococcus pneumoniae's resistance to penicillin (Stephan & Henneberg, 2001). This is what is known as the “Red Queen Effect” where competing selective pressures continually force adaptations in both organisms so that they are in an everlasting race to increased adaptation against the other (Ridley, 2003). Because of this, the increased genetic load (reduction in mean fitness of a population compared to maximum fitness) and the increased fitness of pathogens present the possibility that human survival may be at great risk if medical treatment becomes ineffective (Stephan & Henneberg, 2001).
The influence of biomedicine on the human genome is a prominent factor of the continual evolution of modern humans, perhaps more so if not equal to the previously discussed adaptive mutations. The increase in harmful and non-adaptive deleterious alleles due to medical intervention is a driving force in changing allele frequencies, therefore part of the continuing process of evolution.
Discussion
These examples show considerable evidence in support of the idea that modern humans are still evolving on a genomic level. The ability of the human genome to create mutations and the different phenotypic traits exhibited by these mutations provide the basis for human adaptability. When these differential adaptations are applied to environmental stressors, the alleles that confer the beneficial trait will increase in frequency. While many of these genetic adaptations are in the beginning phases of research and may show conflicting results, there is still ample evidence for evolution based on the adaptations to endemic diseases, environmental stressors, and the consumption of milk for nutritional value.
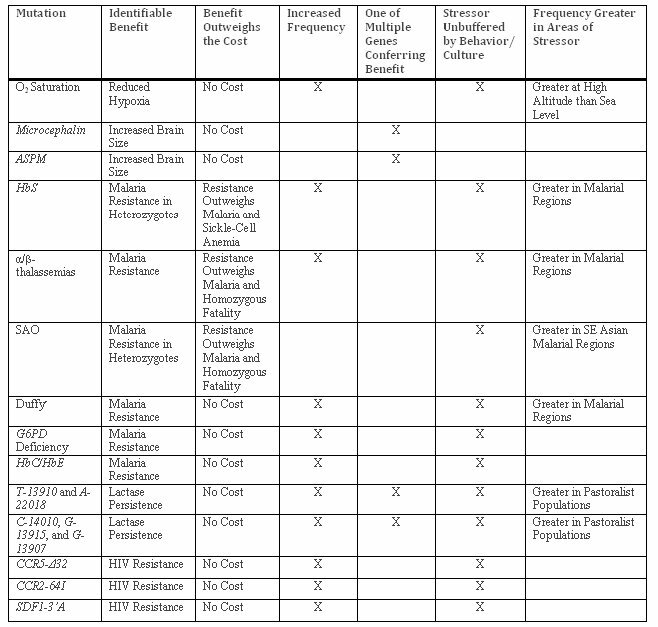
Table 2: Adaptive Mutations Subjected to Criteria for Determining Evidence of Evolution
The diseases associated with high altitude hypoxia (acute mountain sickness and high-altitude pulmonary and cerebral edema) have made it adaptive for individuals with high oxygen saturation of hemoglobin to be more fit. Offspring of individuals with high oxygen saturation have a lower mortality rate, thus a higher percentage of individuals born with this trait are able to reproduce than those born without it. High oxygen saturation has increased in frequency and conforms to five of the six criteria for evidence of evolution.
The genes associated with brain growth, microcephalin and abnormal spindle-like, microcephaly associated, show conflicting evidence. While the frequency of these genes seems to be increasing, there is some question about their effects on brain size. Both genes adhere to only three of the six criteria for evolution so are not considered evidence of modern human evolution.
In areas of underdevelopment and poor mosquito control, malaria has been unbuffered by behavior or culture. Because of this, numerous genetic adaptations have conferred resistance to the parasitic disease. This has increased the reproductive fitness of those individuals with the resistant traits allowing those alleles to be passed on to future generations. In malarial environments, individuals without adaptive alleles have lower reproductive fitness and have fewer offspring, more of whom will not reproduce. This is a “balanced polymorphism” where a homozygous disadvantage is balanced by a heterozygote advantage (Chotivanich et al., 2002). The malarial resistant benefit of HbAS and HbAE balances the disadvantages of HbAA to malaria susceptibility, HbSS to sickle-cell anemia, and a reduced resistance to malaria with HbEE. Over multiple generations, the benefit of being heterozygous has caused an increase in the frequency of these beneficial genotypes in malarial environments. Since the heterozygote, HbAS, is the beneficial genotype, both the ancestral (HbA) and mutative (HbS) alleles will remain in the population. Per generation, there is only a 50% chance of a heterozygote creating a heterozygote offspring regardless of the genotype mated with (HbAS, HbAA, or HbSS). If mated with another heterozygote, the offspring has a 25% chance of having a homozygote HbAA or HbSS, both of which are non-adaptive. Individuals homozygous for the ancestral allele show no resistance to malaria while individuals homozygous for the mutative allele experience sickle-cell anemia. If mated with a homozygote, HbAA or HbSS, the offspring have a 50% chance of having homozygote offspring of HbAA or HbSS, respectively. The cost of this adaptation is the retention of the non-adaptive genotypes and phenotypes exhibiting reduced fitness.
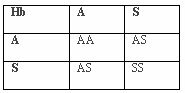
Tables 3, 4, 5. Punnet Squares Showing Offspring Probabilities of Genotype Pairs
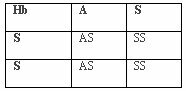
Hemoglobin S, alpha and beta thalassemias, Duffy – blood group, Glucose-6-phosphate dehydrogenase deficiency, Hemoglobin C, and Hemoglobin E all exhibit five of the six required criteria for evolution while South Asian ovalocytosis exhibits four of the six criteria. All of these adaptations to malaria provide sufficient evidence for the evolution of modern humans.
Since multiple alleles conferring lactase persistence have arisen and been favored separately in different geographical regions, and knowing the nutritional benefit of milk, it is clear that humans have evolved this adaptation at a genomic level. The relatively rapid increase in frequency over the last 10,000 years provides a basis for correlating this allele frequency to natural selection. The ability to consume milk provides a nutritional benefit that may not be necessary in developed countries, but provides an important benefit in areas of drought and nutritional stress. Because of this, some human populations have evolved lactase persistence. The alleles T-13910 and A-22018 in Europe and C-14010, G-13915, and G-13907 in Africa all conform to all six of the required criteria for evolution making them prime examples of evidence in support of modern human evolution.
The extremely recent expansion of the HIV virus is the most contemporary selective force for which culture and behavior have not been able to buffer. Regardless of the origin of the CCR5-Δ32 mutation, its protective quality provides an adaptive benefit in HIV exposed individuals. While frequencies of the allele are currently relatively low, except in parts of Europe, it is expected that gene flow and natural selection will spread this allele through areas of HIV exposure. The CCR2-64I and SDF1-3'A alleles also confer resistance to the disease and have reached high frequency at a global level and through Oceania, respectively. The increase in allele frequency, in correlation to high levels of HIV exposure provides sufficient evidence to suggest natural selection and evolution as the cause of these allele frequencies. The three alleles conform to four of the six criteria and are considered evidence of modern human evolution.
The previous examples fit most of, if not all of the required criteria for determining evolution in the human genome. This makes it undeniable that the genome is evolving and the continued presence of these stressors suggests that this evolution is still ongoing. There is no reason to believe that evolution would cease while the stressors are still active, instead these selective forces, as long as they are present, will continue to influence the frequency of alleles for those traits either beneficial or harmful.
In order to prove that modern humans are not evolving, one would have to show that:
- Any alleles increasing in frequency have a low probability of remaining at high frequency,
- Any alleles increasing in frequency due to genetic drift, gene flow, or small population effect exhibit minor, non-directional, or inconsistent genetic changes,
- All stressors can be fully buffered by behavioral or cultural methods, and/or
- The frequency of the mutative allele has no correlation to the presence of the selective force.
Until these criteria can be proven, we must accept the present and continual evolution of the human genome, thus the human species.
Conclusion
While skeptics argue that humans have hit an evolutionary peak and that our culture, medicine and technology have solidified our lack of need for continual evolution, it is medicine and culture that help to solidify continual evolution of the human genome. Culture has been responsible for the evolution agricultural dairying and lactase persistence, responsible for an increase in malaria, thus the evolution of sickle-cell trait, can be said to be a main reason for the rapid spread of HIV, thus is responsible for the evolution of HIV combatant alleles, and is responsible for populations' migration to high altitudes, thus responsible for in increase in high oxygen saturation of hemoglobin. These examples suggest that modern humans remain in a state of constant evolution as a result of common mutation, natural selection, new endemic diseases, and our own cultural influences.
Biographical Summary
I graduated Magna cum Laude with a Bachelors of Science in Anthropology with Departmental Honors from the University of Oregon in 2008. My concentration was in Biological Anthropology and I focused a considerable amount on Evolutionary Theory and Human Evolution and Adaptation.
Acknowledgements
I would like to acknowledge Prof. John R. Lukacs, Ph.D. for advising me on this paper which was my Honors Thesis and Prof. J. Josh Snodgrass, Ph.D. for input and assistance in the early stages of this project.
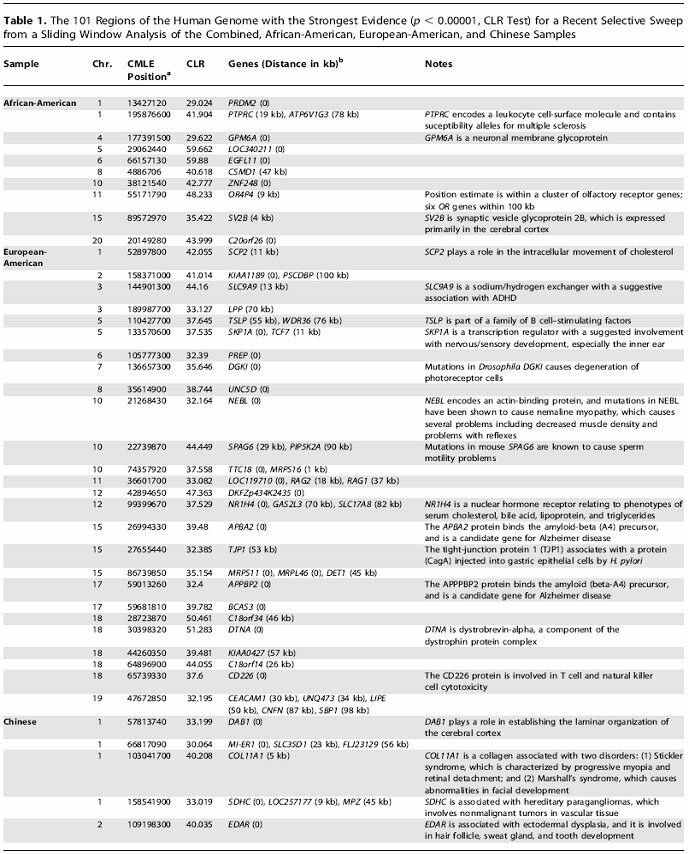
Appendix I

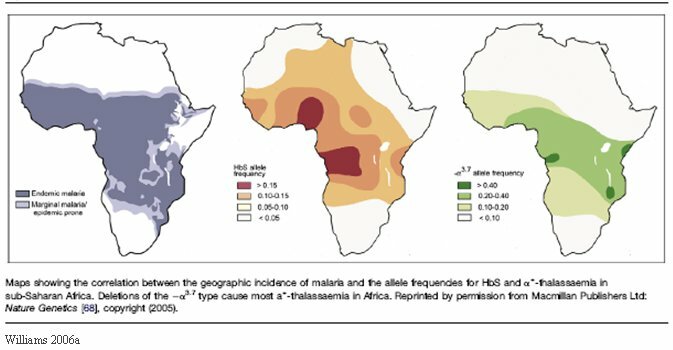
Appendix II

Appendix III
Appendix IV
Endnotes
1. Balter, Michael. 2005a. Are Human Brains Still Evolving? Brain Genes Show Signs of Selection. SCIENCE 309: 1662-1663.
2. Balter, Michael. 2005b. Are Humans Still Evolving? SCIENCE 309: 234-237.
3. Barreiro, Luis B. et al. 2008. Natural Selection Has Driven Population Differentiation in Modern Humans. Nature Genetics 40.3: 340-345.
4. Beall, Cynthia M. 2001. Adaptations to Altitude: A Current Assessment. Annual Reviews Anthropology 30: 423-456.
5. Beall, Cynthia M. et al. 2004. Higher Offspring Survival among Tibetan Women with High Oxygen Saturation Genotypes Residing at 4,000m. PNAS 101.39: 14300-14304.
6. Cappellini, MD & Fiorelli, G. 2008. Glucose-6-Phosphate Dehydrogenase Deficiency. Lancet 371: 64-74.
7. Check, Erica. 2006. How Africa Learned to Love the Cow. NATURE 444.21: 994-996.
8. Chotivanich, Kesinee et al. 2002. Hemoglobin E: A Balanced Polymorphism Protective against High Parasitemias and thus Severe P falciparum Malaria. Blood 100.4: 1172-1176.
9. Cockburn, T Aidan. 1971. Infectious Diseases in Ancient Populations. Current Anthropology 12.1: 45-62.
10. Dobson-Stone, C. et al. 2007. Investigation of MCPH1 G37995C and ASPM A44871G Polymorphisms and Brain Size in a Healthy Cohort. NeuroImage 37: 394-400.
11. Dunbar, R.I.M. & Shultz, Susanne. 2007. Understanding Primate Brain Evolution. Philosophical Transactions of the Royal Society B 362: 649-658.
12. Evans, Patrick D. et al. 2005. Microcephalin, a Gene Regulating Brain Size, Continues to Evolve Adaptively in Humans. SCIENCE 309: 1717-1720.
13. Fairhurst, Rick M. et al. 2003. Aberrant Development of Plasmodium falciparum in Hemoglobin CC Red Cells: Implications for the Malaria Protective Effect of the Homozygous State. Blood 101.8: 3309-3315.
14. Freedman, Scott & Herron, Jon C. 2004. Evolutionary Analysis: Third Edition. New Jersey: Prentice Hall.
15. Galvani, Alison P. & Novembre, John. 2005. The Evolutionary History of the CCR5-Δ32 HIV-Resistance Mutation. Microbes and Infection 7: 302-309.
16. Gibson, Greg. 2007. Human Evolution: Thrifty Genes and the Dairy Queen. Current Biology 17.8: 295-296.
17. Greenwood, Brian M. et al. 2005. Malaria. Lancet 365: 1487-1498.
18. Hay, Simon I. et al. 2004. The Global Distribution and Population at Risk of Malaria: Past, Present, and Future. The Lancet Infectious Diseases 4: 327-336.
19. Hedrick, Philip W. & Verrelli, Brian C. 2006 'Ground Truth' for Selection on CCR5-Δ32. TRENDS in Genetics 22.6: 293-296.
20. Hollox, Edward. 2005. Genetics of Lactase Persistence- Fresh Lessons in the History of Milk Drinking. European Journal of Human Genetics 13: 267-269.
21. Kate, SL & Lingojwar, DP. 2002. Epidemiology of Sickle-Cell Disorder in the State of Maharashtra. Int J Human Genetics 2.3: 161-167.
22. Kimura, M. et al. 2006. Ovalocytosis without Band 3 Gene 27-bp Deletion and Malaria Infection. Anthropological Science 144: 161-164.
23. Kormondy, EJ & Brown, DE. 1998. Human adaptation to cold and heat (Ch. 7). Fundamentals of Human Ecology. Upper Saddle River: Prentice Hall 131-162.
24. Kwiatkowski, Dominic P. 2005. How Malaria Has Affected the Human Genome and What Human Genetics Can Teach Us About Malaria. American Journal of Human Genetics 77: 171-190.
25. Langhi Jr., Dante M. & Bordin, José Orlando. 2006. Duffy Blood Group and Malaria. Hematology 11.5/6: 389-398.
26. Martinson, Jeremy J. et al. 1997. Global Distribution of the CCR5 Gene 32-Basepair Deletion. Nature Genetics 16: 100-103.
27. McDougall, I et al. 2005. Stratigraphic Placement and Age of Modern Humans from Kibish, Ethiopia. NATURE 433: 733-736.
28. Mekel-Bobrov, Nitzan et al. 2005. Ongoing Adaptive Evolution of ASPM, a Brain Size Determinant in Homo sapiens. SCIENCE 309: 1720-1722.
29. Mielke, JH et al. 2006. The genetic basis of human variation (Ch. 2). Human biological variation. Oxford: Oxford U Press 22-46.
30. Miller, Louis H. et al. 2002. The Pathogenic Basis of Malaria. NATURE 415: 673-679.
31. Nielsen, Rasmus et al. 2007. Recent and Ongoing Selection in the Human Genome. Nature Reviews Genetics 8: 857-868.
32. Rambaut, Andrew et al. 2004. The Causes and Consequences of HIV Evolution. Nature Reviews Genetics 5: 52-61.
33. Reed, Floyd A. & Aquadro, Charles F. 2006. Mutation, Selection, and the Future of Human Evolution. TRENDS in Genetics 22.9: 479-484.
34. Reiche, Edna Maria Vissoci et al. 2006. The Effect of Stromal Cell-Derived Factor 1 (SDF1/CXCL12) Genetic Polymorphism on HIV-1 Disease Progression. International Journal of Molecular Medicine 18: 785-793.
35. Ridley, Matt. 2003. The Red Queen: Sex and Evolution in Human Nature. New York: Harper Perennial.
36. Roberts, David J. & Williams, Thomas N. 2003. Hemoglobinopathies and Resistance to Malaria. Redox Report 8.5: 304-310.
37. Schaafsma, Gertjan. 2007. Lactose and Lactose Derivatives as Bioactive Ingredients in Human Nutrition. International Dairy Journal 18: 458-465.
38. Shastry, Barkur S. 2007. SNPs in Disease Gene Mapping, Medical Drug Development and Evolution. Journal of Human Genetics 52: 871-880.
39. Smith, Michael W. et al. 1997. Contrasting Genetic Influence of CCR2 and CCR5 Variants on HIV-1 Infection and Disease Progression. SCIENCE 277: 959-965.
40. Stephan, C.N., & Henneberg, M. 2001. Medicine May Be Reducing the Human Capacity to Survive. Medical Hypotheses 57.5: 633-637.
41. Su, Bing et al. 2000. Distribution of Three HIV-1 Resistance-Conferring Polymorphisms (SDF1-3'A, CCR2-64I, and CCR5-Δ32) in Global Populations. European Journal of Human Genetics 8: 975-979.
42. Tchuenche, Jean M. 2005. Realistic Patterns of Inheritance of Sickle-Cell Anemia Gene: A Theoretical Approach. Journal of Biological Systems 13.1: 13-22.
43. Tishkoff, Sarah A. et al. 2007. Convergent Adaptation of Human Lactase Persistence in Africa and Europe. Nature Genetics 39.1: 31-40.
44. West, John B. 2004. The Physiologic Basis of High-Altitude Diseases. Annals of Internal Medicine 141: 789-800.
45. Wiesenfeld, Stephen L. 1967. Sickle-Cell Trait in Human Biological and Cultural Evolution. SCIENCE 157: 1134-1140.
46. Williams, Thomas. 2006a. Human Red Blood Cell Polymorphisms and Malaria. Current Opinion in Microbiology 9: 388-394.
47. Williams, Thomas. 2006b. Red Blood Cell Defects and Malaria. Molecular and Biochemical Parasitology 149: 121-127.
48. Williamson, Scott H. et al. 2007. Localizing Recent Adaptive Evolution in the Human Genome. PLoS Genetics 3.6: 901-916.